

- 달력에 기능 추가하기 2026.04.21
- 제 2차 씨본 컬러 시뮬레이터 보수작업 2026.04.16
- 백준 섭종공지 2026.04.15
- clinVar EDA를 Polars로 해보자-Appendix. 각 변이별 일짱 2026.04.15
- clinVar EDA를 Polars로 해보자-Pathogenic EDA 2026.04.14
- clinVar EDA를 Polars로 해보자 2026.04.13
- Polars 데이터프레임도 시각화가 되나요? 2026.04.10
- Polars를 써보자 2026.04.09
- Medical Cost Personal Datasets 2026.03.18
- Red Wine Quality 2026.03.13
- Palmer Archipelago (Antarctica) penguin data 2026.03.11
- 웹 프로젝트를 깃헙에 올릴 수 있다? 뿌슝빠슝 2026.03.08
- 포폴 웹페이지화-더 이상의 자세한 설명은 생략한다 2026.03.08
- 포폴 웹페이지화-뼈대 잡기 2026.03.07
- 개 얼탱이 없는 작업이 온다 두둥 2026.03.06
아 EDA 할 생각 없냐고요?
맥북 반납해서 EDA를 못합니다... 리눅스 노트북은 데이터 분석 돌리면 뻗어서 평일에 글쓸때나 자바스크립트 할 때 말고 켜본적 없음...

그새 글꼴이 많이 바뀌긴 했는데... 그래서 이 달력에 뭔 기능을 추가할거임? 바로 오늘 날짜로 되돌아가는 기능과 몇년 몇월로 이동하는 기능이다. 전자는 뭔지 알겠는데 후자는 뭐임? 여러분 이 달력에서 1991년 3월로 어떻게 가는지 아십니까? 1991년 3월 될때까지 이전달을 급나 눌러야됩니다. 일단 햇수로만 36년인데 1년이 12개월이니까 대충 클릭질 몇 번 해야 하는지 견적이 나오시죠?
오늘 날짜로 돌아가는 버튼
let goTodayButton = document.querySelector('#gotoday');이게 HTML 구조는 간단한데 CSS가… ㅋㅋㅋㅋㅋ 내가 오래전에 한거라 뭘 어떻게 한건지 헷갈림… 여러분은 이런 사태를 미연에 방지하실거면 주석 다십쇼.
아무튼 갖고옵시다.
// 오늘 날짜로 고고싱하는 버튼
goTodayButton.addEventListener('click', (e) => {
// 1. 전역 변수인 'date'를 현재 시점의 날짜로 갱신
date = new Date();
// 2. 갱신된 date 객체를 바탕으로 달력을 다시 그림
renderCalendar();
});사실 원리는 간단합니다. 오늘 날짜로 객체 생성하고 달력 다시 그리면 돼요. 그러면 일단 오늘 날짜로 복귀는 됐고… 이제 특정 년도 특정 달로 이동하는 기능 만들자.
특정 날짜로 이동하기
// 특정 날짜로 고고싱하는 버튼
goDateButton.addEventListener('click', (e) => {
let goYear = document.querySelector('#goyear').value;
let goMonth = document.querySelector('#gomonth').value;
// 1. 입력값이 있는지 확인 (비어있으면 동작 안 함)
if (goYear && goMonth) {
// 2. 전역 변수인 date 객체의 연도와 월을 업데이트
// 숫자로 형변환(Number)해주고, 월은 반드시 -1을 해줍니다.
date.setFullYear(Number(goYear));
date.setMonth(Number(goMonth) - 1);
// 날짜가 해당 월의 마지막 날을 넘어가는 버그를 방지하기 위해 1일로 세팅하는 것이 안전합니다.
date.setDate(1);
// 3. 갱신된 정보를 바탕으로 달력 다시 그리기
renderCalendar();
} else {
alert("이동할 연도와 월을 모두 입력해주세요!");
}
});이것도 걍 그 날짜로 달력 다시 그리면 되던데요? 근데 날짜는 왜 1일로 바꿔서 그렸나요? 어차피 우리는 그 특정 해 특정 월로 이동만 할 거잖아요. 그리고 이 달력을 쓰는 당일 날짜가 31일이면 말이이 30일인 달이나 2월로 이동할때 날짜 안 바꾸면 2월 31일은 뭐야!!! 이러고 컴퓨터가 밥상 뒤집어요.

이렇게 하면 일단은 되는'데'… 아직 끝나지 않은 문제가 있다.
콘솔에만 뜨는 메시지 처리하기
여러분들에게는 보이지 않지만, 사실 이 달력을 구동할 때 콘솔창에 보이는 메시지가 하나 있다.

바로 이건데… 저 달력을 만들때 아마 채찍피티가 없었거나 초창기라 디버깅을 못했을거다. 근데 지금은 채찍피티 클로드 에미나이가 다 있잖아요? 그래서 디버깅 해달라고 할거임.

그렇다고 합니다. 그러니까 기존 코드에서는 함수에서 자체적으로 범위를 체크하고 있었는데, 이것때문에 혼선이 와서 저런 메시지가 뜨는 것이다. 그러니까 걍 자체 체크하는걸 없애고 Date 객체가 알아서 보정하게 두면 된다 이거죠.
function dayCalcDisplay(startYear, startMonth, startDay) {
// 1. 넘겨받은 연, 월, 일을 바탕으로 실제 Date 객체 생성
// (월은 0~11이므로 -1, 일은 그대로)
// 예: 2025년 2월의 이전달 날짜로 '31'이 들어와도
// new Date(2025, 1, 31)은 자동으로 2025년 3월 3일로 인식됩니다.
var tempDate = new Date(startYear, startMonth - 1, startDay);
// 2. 보정된 진짜 연/월/일을 다시 추출
var realYear = tempDate.getFullYear();
var realMonth = tempDate.getMonth() + 1;
var realDay = tempDate.getDate();
// 3. (옵션) 윤년 체크 로직은 사실 lunarCalc 내부에서 수행하므로
// 여기서는 범위 체크를 삭제하거나 보정된 값을 사용합니다.
if (realYear < 1900 || realYear > 2040) {
return ""; // 지원 범위 밖이면 빈칸 처리
}
/* 양력/음력 변환 (보정된 진짜 날짜를 넣음) */
var lunar = lunarCalc(realYear, realMonth, realDay, 1);
return (lunar.leapMonth ? "윤" : "") + lunar.month + "." + lunar.day;
}그래서 함수가 더 짧아졌음. 근데 저거 생각해보니까 2040년 넘어가면 ""준다고? 아니… 그때 나 50살인데? 뭐 그래요… 그때쯤 되면 뭔가 새로운 방법이 나오겠지… 그때 코드를 바꾸던가 합시다…
'Coding > JavaScript' 카테고리의 다른 글
| 제 2차 씨본 컬러 시뮬레이터 보수작업 (0) | 2026.04.16 |
|---|---|
| 포폴 웹페이지화-더 이상의 자세한 설명은 생략한다 (0) | 2026.03.08 |
| 포폴 웹페이지화-뼈대 잡기 (0) | 2026.03.07 |
| 개 얼탱이 없는 작업이 온다 두둥 (0) | 2026.03.06 |
| 씨본 팔레트 컬러 시뮬레이터 보수작업 (0) | 2026.02.22 |
내가 이걸 또 하게 되다니… 싶었는데 쓰다가 또 불편한 게 나와서 에미나이 도움 받아서 기능을 추가했다.

이거 보여요? 기존에는 없었던 저 HEX 코드들. 이게 중간색을 쓰고 싶은데 #rrggbb 코드가 없으니까 내가 개발자도구 열고 들어가서 변환을 해야되는데 이게 증말 번거롭습니다… 그리고 나야 개발자도구의 존재를 안다지만 모든 사람들이 그렇진 않잖아요?
for (let i = 0; i < n; i++) {
const step = i / (n - 1);
const r = Math.round(startRgb[0] + (endRgb[0] - startRgb[0]) * step);
const g = Math.round(startRgb[1] + (endRgb[1] - startRgb[1]) * step);
const b = Math.round(startRgb[2] + (endRgb[2] - startRgb[2]) * step);
const toHex = (c) => c.toString(16).padStart(2, '0');
const hexCode = `#${toHex(r)}${toHex(g)}${toHex(b)}`.toUpperCase();
const chip = document.createElement('div');
chip.classList.add('palette_chip');
chip.style.backgroundColor = `rgb(${r}, ${g}, ${b})`;
chip.style.flex = "1";
// 스타일 및 텍스트 설정
chip.style.display = "flex";
chip.style.alignItems = "center";
chip.style.justifyContent = "center";
chip.style.cursor = "pointer"; // 클릭 가능하다는 표시
chip.style.color = (r * 0.299 + g * 0.587 + b * 0.114) > 186 ? '#000' : '#fff';
chip.innerText = hexCode;
chip.onclick = () => {
navigator.clipboard.writeText(hexCode).then(() => {
// alert 대신 토스트 호출!
showToast(`${hexCode} copied!`);
}).catch(err => {
console.error('복사 실패:', err);
});
};
palette_div.appendChild(chip);
}저정도 되면 어디까지가 한 블록인지 헷갈려요... const toHex = (c) => c.toString(16).padStart(2, '0');랑 const hexCode = `#${toHex(r)}${toHex(g)}${toHex(b)}`.toUpperCase(); 보여요? 저걸로 10개 색상을 HEX코드로 변환한 다음에 이너텍스트 박을거고, 그 네모를 누르면 HEX 코드가 복사되면서 복사됐음! 도 할 거다. 처음에는 alert였는데 alert는 확인버튼을 눌러야되거든요? 그래서 토스트 메시지로 바꿨음.
function showToast(message) {
let toast = document.getElementById('toast');
if (!toast) {
toast = document.createElement('div');
toast.id = 'toast';
document.body.appendChild(toast);
}
toast.innerText = message;
toast.classList.add('show');
// 2초 뒤에 사라지게 설정
setTimeout(() => {
toast.classList.remove('show');
}, 2000);
}그래서 토스트 창(그 있어요 나왔다가 들어가는 창) 생성 함수가 추가됐고
/* 토스트 메시지 기본 스타일 */
#toast {
position: fixed;
bottom: 30px;
left: 50%;
transform: translateX(-50%);
background-color: rgba(0, 0, 0, 0.8);
color: #fff;
padding: 12px 24px;
border-radius: 8px;
font-size: 14px;
opacity: 0;
transition: opacity 0.3s, bottom 0.3s;
z-index: 9999;
pointer-events: none; /* 클릭 방해 금지 */
}
/* 토스트가 보일 때의 상태 */
#toast.show {
opacity: 1;
bottom: 50px;
}관련 CSS도 추가됐습니다. HTML은 바뀐거 없음.
'Coding > JavaScript' 카테고리의 다른 글
| 달력에 기능 추가하기 (0) | 2026.04.21 |
|---|---|
| 포폴 웹페이지화-더 이상의 자세한 설명은 생략한다 (0) | 2026.03.08 |
| 포폴 웹페이지화-뼈대 잡기 (0) | 2026.03.07 |
| 개 얼탱이 없는 작업이 온다 두둥 (0) | 2026.03.06 |
| 씨본 팔레트 컬러 시뮬레이터 보수작업 (0) | 2026.02.22 |
https://www.acmicpc.net/board/view/165799
내가 코딩을 시작하면서부터 풀어왔던 백준이 섭종 공지를 때렸다. 오호 통재라.
나는 다른 사이트 말고 오직 여기만 이용했었다. 최근에는 이차원 배열에서 막혔지만, 적어도 이것만큼은 AI의 힘을 빌리고 싶지 않아서, 그리고 부트캠프 하느라 바쁨을 핑계로 안 했었다. 백준이 언제까지나 거기까지 있을 줄 알고 말이지.
나중에 복각해주세요. 그때까지 저는 영원한 실버 1로 남겠습니다.
'Coding > 코딩잡담' 카테고리의 다른 글
| 웹 프로젝트를 깃헙에 올릴 수 있다? 뿌슝빠슝 (0) | 2026.03.08 |
|---|---|
| 안티그래비티를 써보았다. (0) | 2025.12.19 |
| CONNECT BY와 계층형 질의 (0) | 2025.11.14 |
| 스키마 (0) | 2025.11.14 |
| Select문에도 순서가 있다 (0) | 2025.11.13 |
그 전에 clinVar 하면서 태블로 대시보드 만들었죠? 거기서 염색체별로 Top 25 만들고 그랬는데… 그거를 이제 CLNVC(변이)별로 뭐가 제일 많은지 보자는 얘기다.
SNV는 저번 글에 나와있었기 때문에 생략.
들어가기 전에-이게 뭔 변이임?
이 블로그에 들어오시는 분들중에는 생물정보학을 하고 있거나, 나처럼 업으로 삼지는 않았지만 거기에 관심이 있거나, 생물학 전공인 경우도 있겠지만 어때요? 여기 들어와서 clinVar라는 걸 처음 보신 분도 계시지 않습니까? 그래요, 그겁니다. EDA 따라오면서도 이게 뭐여 해서 뭔가 찾아보니 보이는 것은 꼬부랑글씨였으며…
물론 유전자에 문제가 생긴다고 다 질병이 되는 건 아니고, 피부 색이나 눈 색, 머리카락 색같이 사람의 형질이 달라지는 경우도 있긴 있습니다. 근데 여기 부록으로 나온건 다 패소제닉(Pathogenic)이라 뻑나면 병나는거예요.
1. Deletion: 일단 뭔가 나간 변이다. 나간 게 염기일수도 있고, 염색체 일부(염기 뭉텅이)일수도 있다. 그러니까 집 설계도라고 치면 벽이나 문, 창문같은게 하나 빠지거나 방 하나가 나가거나 뭐 그런거.
2. Duplication: 뭔가 1+1이 됐다. 집 설계도에 방이 두개였는데 세개가 된다거나, 창문이 하나였는데 두개가 된다거나… 이게 무조건 위험한 건 아니고 카피 넘버 배리언스(CNV)라고 해서 그 자체가 형질이 되는 경우도 있습니다. 헌팅턴 무도병도 유전자 내에 CAG가 불어나서 생기는거예요.
3. Insertion: 뭔가 껴들었다. 염기가 끼어들 수도 있고 유전자 자체가 끼어들 수도 있는데, Transposon(유전자가 이사다님)으로 인해 유전자가 끼어들어서 생기는 현상중에 유명한건 얼룩덜룩한 옥수수.
4. 1+3. 뭐가 드나드는게 인델이다. 인서션 딜리션 해서 인델.
5. Inversion: 유전자가 반대로 들어갔다. 그니까 집 설계도로 치자면 문이 밖으로 열려야되는데 안으로 열리게 들어간거다. 물론 유전자가 반대로 들어가는건 방...에 비유할수가... 있어요? 방이 위아래 거꾸로 들어갈 수가 있음?
6. Microsatellite: 짤막한 염기서열이 반복되는 돌연변이. 이것도 뭐 사람들한테 위험하다… 이런 건 아니고… 맞죠? 전공수업때는 못봤던거임…
7. Variation: 찾아봤더니 사전적인 정의가 나온다…
Deletion
Duplication

Insertion

Indel

Y랑 미토콘드리아는 걍 없나본데...?
Inversion

Microsatellite

Variation

'Coding > EDA' 카테고리의 다른 글
| clinVar EDA를 Polars로 해보자-Pathogenic EDA (0) | 2026.04.14 |
|---|---|
| clinVar EDA를 Polars로 해보자 (0) | 2026.04.13 |
| Medical Cost Personal Datasets (0) | 2026.03.18 |
| Red Wine Quality (0) | 2026.03.13 |
| Palmer Archipelago (Antarctica) penguin data (0) | 2026.03.11 |
지난 이야기: 아 염색체별로 CLNSIG 비중이 이렇구나
Pathogenic 일로와봐
pathogenic_df = clinvar_df.filter(pl.col('CLNSIG_Group') == 'Pathogenic')이렇게 하면 됩니다.
CLNVC별로 보기
clnvc_grp = clinvar_df.group_by('CLNVC').agg(
pl.col('CLNSIG').count().alias("Total")
).sort('Total', descending=True)묶어드렸습니다^^
fig = go.Figure()
fig.add_trace(
go.Bar(x = clnvc_grp['CLNVC'], y = clnvc_grp['Total'], marker_color = px.colors.sequential.algae, text = clnvc_grp['Total'])
)
fig.update_layout(
width = 1200, height = 800,
xaxis=dict(title='Pathogenic Variant Count', tickangle = -90),
yaxis=dict(title='Count'),
title = "ClinVar Group Distribution",
margin = dict(t=50, l=10, r=10, b=10)
)
# fig.write_image('pathogenic_distributuion.png')
fig.show()
SNV가 너무 압도적인데...?
fig = px.treemap(
clnvc_grp,
path = ['CLNVC'],
values = 'Total',
color = 'Total',
color_continuous_scale = 'algae',
title = "Pathogenic Variant Distribution",
)
# text_auto 대신 update_traces를 사용하여 블록 안에 텍스트를 강제로 넣습니다.
fig.update_traces(
# label(그룹명)과 value(숫자)를 함께 표시하라는 명령입니다.
textinfo = "label+value",
# 숫자 포맷을 지정합니다 (천 단위 콤마: ,d)
texttemplate = "%{label}<br>%{value:,d}",
# 텍스트가 블록 크기에 맞게 조절되도록 설정
textfont_size = 20,
insidetextfont_family = "NanumSquare_ac" # 설정하신 폰트 적용
)
fig.update_layout(
width = 1200,
height = 800,
margin = dict(t=50, l=10, r=10, b=10)
)
# fig.write_image('pathigenic_distributuion_heatmap.png')
fig.show()
SNV 면적이 압도적이죠?
fig = go.Figure()
fig.add_trace(go.Pie(
labels = clnvc_grp['CLNVC'],
values = clnvc_grp['Total'],
textinfo = 'label+percent', # 이름과 퍼센트 동시에 표시
insidetextorientation = 'radial',
marker = dict(colors=px.colors.qualitative.Vivid) # 전역 설정 색감 유지
))
fig.update_layout(
title_text = "Pathogenic Variant Distribution", title_x=0.9,
width = 1200,
height = 800,
margin = dict(t=50, l=20, r=20, b=10)
)
# fig.write_image('pathgenic_distributuion_pie.png')
fig.show()
음… 이거 포폴에 써먹을 수 있나…
상염색체가 쪽수가 많은 건 알겠는데, 뭐가 제일 많음?
# 1. 데이터에 있는 실제 값 순서 (가출 방지용)
real_values = ["Sex_Chrom", "Autosome", "Mitochondria"]
# 2. 그래프 위에 표시하고 싶은 예쁜 제목들 (순서 일치 필수!)
display_titles = ["Sex Chromosome", "Autosome", "Mitochondria"]
# 3. 서브플롯 생성
fig = make_subplots(
rows=1, cols=3,
subplot_titles=display_titles, # 여기서 예쁜 제목 적용!
horizontal_spacing=0.08
)
# 4. 반복문은 실제 값(real_values)으로 돌립니다
for i, real_val in enumerate(real_values):
curr_df = clnvc_grp.filter(pl.col('CHROM_Type') == real_val)
# 내림차순 정렬해서 보기 좋게
curr_df = curr_df.sort("Total", descending=True)
fig.add_trace(
go.Bar(
x = curr_df['CLNVC'],
y = curr_df['Total'],
text = curr_df['Total'],
texttemplate = '%{y:,}',
textposition = 'outside',
# 색깔은 이미 설정한 대로!
marker_color = px.colors.sequential.algae[-(i*3+1)]
),
row = 1, col = i + 1
)
# 5. 레이아웃 정리
fig.update_layout(
height=600,
width=1250,
title_text="ClinVar Distribution by Chromosome Type",
showlegend=False,
margin=dict(t=100, l=20, r=20, b=50), # 제목 공간 확보 위해 t(top) 늘림
)
# fig.write_image('pathgenic_chr_distributuion_subplot.png')
fig.show()
그냥 SNV가 압도적이다.
chr_order = [str(i) for i in range(1, 23)] + ['X', 'Y', 'MT']
# 2. 비율 계산
clnsig_perc = clnvc_grp.with_columns(
(pl.col("Total") / pl.col("Total").sum().over("CHROM") * 100).alias("Percentage")
)
# 3. Plotly Express로 그리기
fig = px.bar(
clnsig_perc,
x = "CHROM",
y = "Percentage",
color = "CLNVC",
text = "Percentage",
title = "CLNVC ratio by Chromosome",
color_discrete_sequence = px.colors.qualitative.Vivid,
# --- 이 부분이 마법의 한 줄입니다 ---
category_orders = {"CHROM": chr_order}
# ----------------------------------
)
fig.update_traces(
texttemplate='%{text:.1f}%',
textposition='inside',
insidetextfont_family="NanumSquare_ac" # 설정하신 폰트 적용
)
fig.update_layout(
width=1250,
height=800,
barmode='stack',
xaxis=dict(title='Chromosome'),
yaxis=dict(title='Percentage (%)'),
margin=dict(t=100, l=20, r=20, b=50),
)
# fig.write_image('chr_ratio.png')
fig.show()
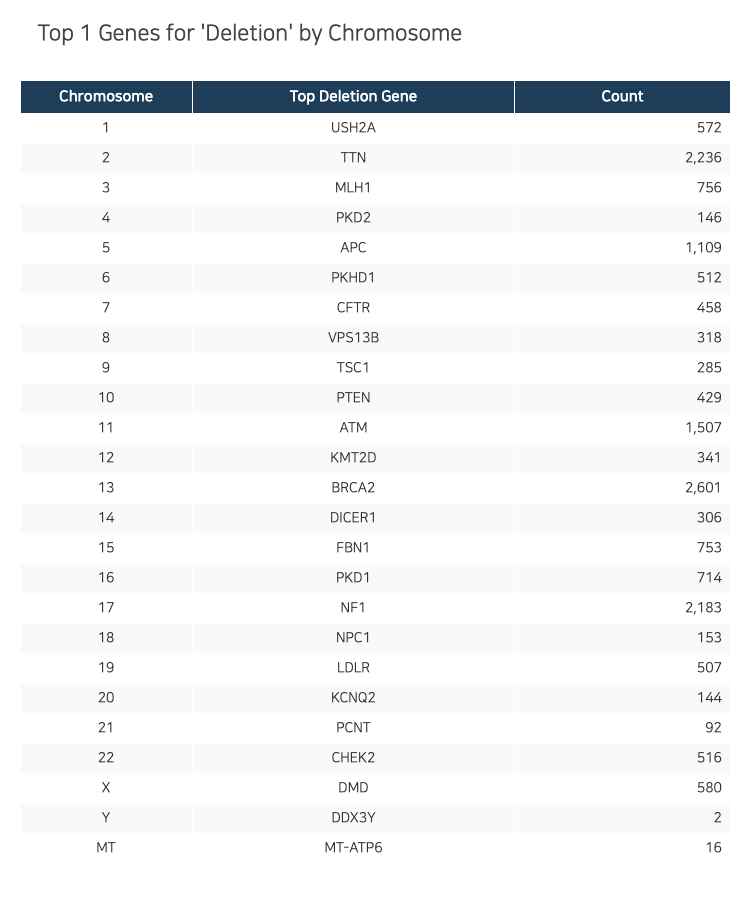
저게 문자로 잡혀서 순서 수기로 잡아줘야 합니다. 아무튼 각 염색체별로 제일 많은 건 SNV고 그 다음에 Deletion이다. 미토콘드리아의 경우 Deletion 다음으로 Insertion, 나머지 염색체는 Deletion 다음으로 Duplication이 제일 많다.
# 1. 염색체 정렬 순서 정의
chr_order = [str(i) for i in range(1, 23)] + ['X', 'Y', 'MT', 'M']
# 순서대로 숫자를 매칭한 딕셔너리 생성 (1:0, 2:1, ..., X:22, Y:23...)
chr_map = {val: i for i, val in enumerate(chr_order)}
# 2. 데이터 집계 및 필터링
top_gene_df = (
pathogenic_df
.group_by(['CHROM', 'GENE_SYMBOL'])
.agg(pl.count('GENE_SYMBOL').alias("Count"))
.filter(pl.col("Count") == pl.col("Count").max().over("CHROM"))
.unique(subset=["CHROM"], keep="first")
)
# 3. 정렬 로직 수정 (최신 Polars 버전 대응)
top_gene_df = (
top_gene_df
.with_columns(
pl.col("CHROM").cast(pl.Utf8)
)
.with_columns(
# replace 대신 replace_strict를 사용하고, 매핑 안 되는 값은 99로 처리합니다.
pl.col("CHROM").replace_strict(chr_map, default=99).alias("sort_idx")
)
.sort("sort_idx")
)위에도 썼지만 X, Y가 껴서 문자라 수동으로 정렬해줘야 합니다... 아무튼 각 염색체별로 변이가 가장 많은 유전자를 갖고왔다. 판다스였으면 idxmax 쓰는건데 까비...
# 1. 표에 들어갈 데이터 정리 (위에서 정렬된 top_gene_df 활용)
header_values = ["<b>Chromosome</b>", "<b>Top Gene Symbol</b>", "<b>Variant Count</b>"]
cell_values = [
top_gene_df["CHROM"],
[f"<b>{gene}</b>" for gene in top_gene_df["GENE_SYMBOL"]], # 유전자 이름 강조
[f"{count:,}" for count in top_gene_df["Count"]] # 천 단위 콤마
]
# 2. Plotly Table 생성
fig = go.Figure(data=[go.Table(
# 컬럼 너비 비율 조절
columnwidth = [100, 150, 120],
# 헤더 스타일
header = dict(
values = header_values,
fill_color = '#2E4A3F', # Algae 계열의 짙은 색
align = 'center',
font = dict(color='white', size=16, family="NanumSquare_ac"),
height = 40
),
# 셀 스타일
cells = dict(
values = cell_values,
fill_color = ['#F5F5F5', 'white'], # 줄바꿈 배경색 효과
align = ['center', 'center', 'right'],
font = dict(color='#333', size=14, family="NanumSquare_ac"),
height = 30
)
)])
# 3. 레이아웃 설정
fig.update_layout(
title = "Top Mutated Gene by Chromosome (ClinVar Pathogenic)",
width = 800,
height = 900, # 데이터가 24줄(1~22, X, Y, M) 정도 되니 높이를 넉넉하게
margin = dict(t=80, l=20, r=20, b=20)
)
# fig.write_image('chr_gene.png')
fig.show()
그… 여러분… Plotly에서는 표를 그릴 수 있습니다. 뭐 마우스 올려도 뭐 없긴 한데 아무튼.

근데 이런것도 될 줄은 몰랐지… 이게 뭔 표임? 각 염색체별로 변이가 가장 많은 유전자들만 꼽아서 그 중에서 1, 2, 3등을 매긴 결과다. BRCA2, TTN, BRCA1 순으로 변이가 많다. 근데 저 SRY는 뭐죠? 성별 결정하는 유전자입니다. 찾아보니 고환 결정 인자란다.
# 1. 염색체 정렬 순서 정의
chr_order = [str(i) for i in range(1, 23)] + ['X', 'Y', 'MT', 'M']
# 순서대로 숫자를 매칭한 딕셔너리 생성 (1:0, 2:1, ..., X:22, Y:23...)
chr_map = {val: i for i, val in enumerate(chr_order)}
# 2. 데이터 집계 및 필터링
top_gene_df = (
pathogenic_df
.group_by(['CHROM', 'CLNVC','GENE_SYMBOL'])
.agg(pl.count('GENE_SYMBOL').alias("Count"))
.filter(pl.col("Count") == pl.col("Count").max().over("CHROM"))
.unique(subset=["CHROM"], keep="first")
)
# 3. 정렬 로직 수정 (최신 Polars 버전 대응)
top_gene_df = (
top_gene_df
.with_columns(
pl.col("CHROM").cast(pl.Utf8)
)
.with_columns(
# replace 대신 replace_strict를 사용하고, 매핑 안 되는 값은 99로 처리합니다.
pl.col("CHROM").replace_strict(chr_map, default=99).alias("sort_idx")
)
.sort("sort_idx")
)# 1. 변이 수 기준으로 데이터 정렬 (표 상단에 TOP 3가 오게 하려면 여기서 정렬)
# 현재 top_gene_df가 염색체 순서라면, 강조 로직을 수치 기준으로 적용해야 합니다.
top_gene_df = top_gene_df.sort("Count", descending=True)
# 2. 행별 배경색 결정 함수
def get_rank_color(i):
if i == 0: return '#FFD700' # Gold (1위: BRCA2)
if i == 1: return '#E5E4E2' # Platinum/Silver (2위: TTN)
if i == 2: return '#CD7F32' # Bronze (3위: BRCA1)
return 'white' if i % 2 == 0 else '#F9F9F9' # 나머지 가독성용 줄무늬
row_colors = [get_rank_color(i) for i in range(len(top_gene_df))]
# 3. 데이터 구성
header_values = ["<b>Chromosome</b>", "<b>CLNVC</b>", "<b>Top Gene Symbol</b>", "<b>Variant Count</b>"]
cell_values = [
top_gene_df["CHROM"],top_gene_df["CLNVC"],
[f"<b>{g}</b>" if i < 3 else g for i, g in enumerate(top_gene_df["GENE_SYMBOL"])],
[f"{c:,}" for c in top_gene_df["Count"]]
]
# 4. Table 생성
fig = go.Figure(data=[go.Table(
columnwidth = [80, 150, 100],
header = dict(
values = header_values,
fill_color = '#2E4A3F', # 헤더는 짙은 Algae색 유지
align = 'center',
font = dict(color='white', size=16, family="NanumSquare_ac")
),
cells = dict(
values = cell_values,
fill_color = [row_colors] * 3,
align = ['center', 'center', 'right'],
# 글자색은 무조건 검은색 계열로 고정하여 가시성 확보
font = dict(color='#333', size=14, family="NanumSquare_ac"),
height = 35,
line_color = '#E5E5E5' # 셀 구분선 살짝 추가
)
)])
fig.update_layout(
title = "Top 3 Mutated Genes Highlighted",
width = 750,
height = 1010,
margin = dict(t=80, l=20, r=20, b=20)
)
# fig.write_image('chr_gene_clnvc.png')
fig.show()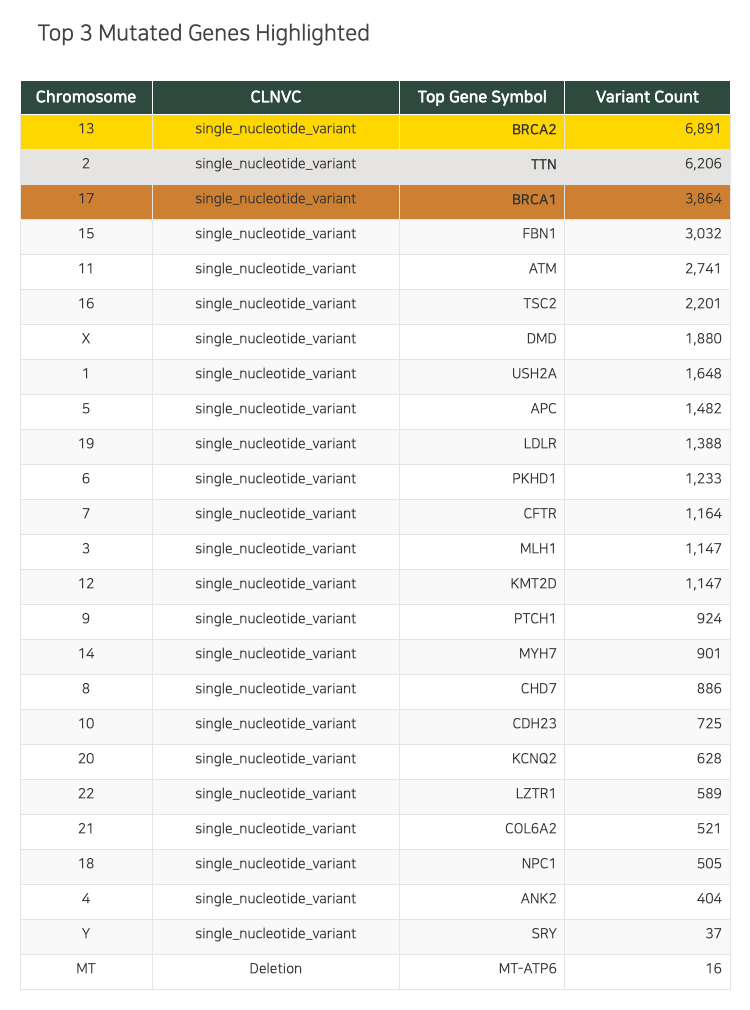
보시면 미토콘드리아(MT-ATP6) 빼고 다 SNV죠? 저게 개인적으로는 나비효과라고 생각하는데, 염기 하나가 바뀐 것 때문에 단백질이 바뀌고, 그걸로 인해서 기능에 문제가 생기는 게 질병으로 이어지기 때문이다. 그러니까 아주 작은 염기 하나가 바뀌는 것 때문에, 염기보다 훨배 큰 사람이 영향을 받는다 이거지.

반대로 변이가 가장 적은 유전자인데... 이게 1? 왜 1이죠? 일단 이렇다 하고 단정지을 수는 없는데... 치사유전 아세요? 혈우병이 치사유전이잖아요. 여자의 경우 혈우병 유전자를 양쪽 염색체가 둘 다 갖고있으면 보통은 사산됩니다. 물론 생물학에는 100%가 없으니 그 와중에도 정말 드문 확률로 살아서 나올 가능성은 있지만...
웨스턴블록 해본 사람들은 표적 단백질 말고 컨트롤? 그런거 같이 매겨서 밴드 굵기 보셨을텐데 이걸 왜 볼까요? 그죠. 이거 발현 안되면 X되는 단백질이라 보는겁니다. 풀때기에 엽록체가 없다고 생각해보세요. 광합성을 못해서 멀리 가십니다.
아니 왜 갑자기 웨스턴 얘기가 나옴? 나도 100% 그렇다고는 못 하지만, 저 유전자들은 변이가 생기면 질병 생기는 수준이 아니라 그냥 죽거나(생존을 못하거나) 대를 못 이을 가능성이 있습니다.
'Coding > EDA' 카테고리의 다른 글
| clinVar EDA를 Polars로 해보자-Appendix. 각 변이별 일짱 (0) | 2026.04.15 |
|---|---|
| clinVar EDA를 Polars로 해보자 (0) | 2026.04.13 |
| Medical Cost Personal Datasets (0) | 2026.03.18 |
| Red Wine Quality (0) | 2026.03.13 |
| Palmer Archipelago (Antarctica) penguin data (0) | 2026.03.11 |
전에 깔짝깔짝 판다스랑 비교했던 폴라스로… EDA가 될지 해봤다. 그래서 전에 했던거랑 내용은 같은데, 비교하는 툴이 달라지는겁니다. 이거 아마 포폴에도 폴라스 플롯틀리로 올라갈듯함. 근데 새로 나온건 알겠어, 이걸 써봐야 해? 네카라쿠배의 배에서 쓴답니다.
전처리는 이전 과정이랑 비슷하니까 그룹바이랑 필터 위주로 ㄱㄱ합시다.
clinvar_df = pl.read_csv('data/clinvar_20260404.csv', infer_schema_length=0)얘는 판다스에서 붙는 메모리 관련 옵션이 아예 안 붙는다. 근데 뭔가 붙어있지 않냐고? 걍 열면 Original error: invalid primitive value found during CSV parsing 에러 뜨니까 걍 다 읽고 판별하셈 한 겁니다. 그렇게 해도 1초 좀 넘게 걸려요.
CLNSIG으로 묶기
이게 일단 돌리는 방식은 예전이랑 동일한데, 데이터가 달라져서(4월 4일자껄로 진행) 결과가 달라질 수 있다. 4월 4일자에 든 게 4,403,603개고 전에 했던건 좀 적게 들어있었음... 그래서 총계 이런거에서는 차이가 날 수밖에 없음.
clinvar_df.group_by('CLNSIG_Group').agg(
pl.col('CLNSIG').count().alias("Total")
)놀라운 사실을 하나 알려주지면, Polars에서는 단순히 그룹바이가 되는 걸 떠나서 .agg로 집계 매긴 다음 그 집계 칼럼에 별칭을 붙일 수 있다. 판다스도 되긴 되겠지만 쟤는 한번에 저게 된다. 두번째줄 pl.col('CLNSIG').count().alias("Total")가 무슨 의미냐면 CLNSIG 칼럼으로 묶고 셀 건데 그 결과물 칼럼 이름을 Total로 해달라는 의미. SQL 해보셨으면 감이 좀 왔을 것이다.
당연한 얘기지만, alias 없어도 저 코드는 작동을 한다. 근데 어지간하면 있는 편이 좋다.

alias 안 주면 Total이 아니라 CLNSIG으로 나오는데 이게 뭔줄 앎? 그럼 이걸로 이제 플롯틀리 그래프를 그리기 전에 정렬을 좀 해야되는데…
clinvar_df.group_by('CLNSIG_Group').agg(
pl.col('CLNSIG').count().alias("Total")
).sort("Total", descending=True) # 정렬을~ 돌려다아아아오~걍 뒤에 소트 붙이시면 알아서 정렬 됩니다. 모든 정렬은 오름차순이 기본이니 내림차순으로 정렬해주면 된다. 이걸 이제 그대로 플롯틀리에 떤지면 안되고… 변수명 할당해서 줘야됨.
fig = go.Figure()
fig.add_trace(
go.Bar(x = clnsig_grp['CLNSIG_Group'], y = clnsig_grp['Total'], marker_color = px.colors.sequential.Cividis,text = clnsig_grp['Total'])
)
fig.update_layout(
width = 1200, height = 800,
xaxis=dict(title='ClinVar Significance Group'),
yaxis=dict(title='Count'),
title = "ClinVar Group Distribution"
)
# fig.write_image('group_distributuion.png')
fig.show()여러분들은 여기서 피똥 쌀 것이다. 일단 나는 여기서 피똥 쌌음... 아니 전역으로 컬러테이블 박아놨는데 마커는 또 일일이 주래요 이게 말이야 당나귀야 세상에...
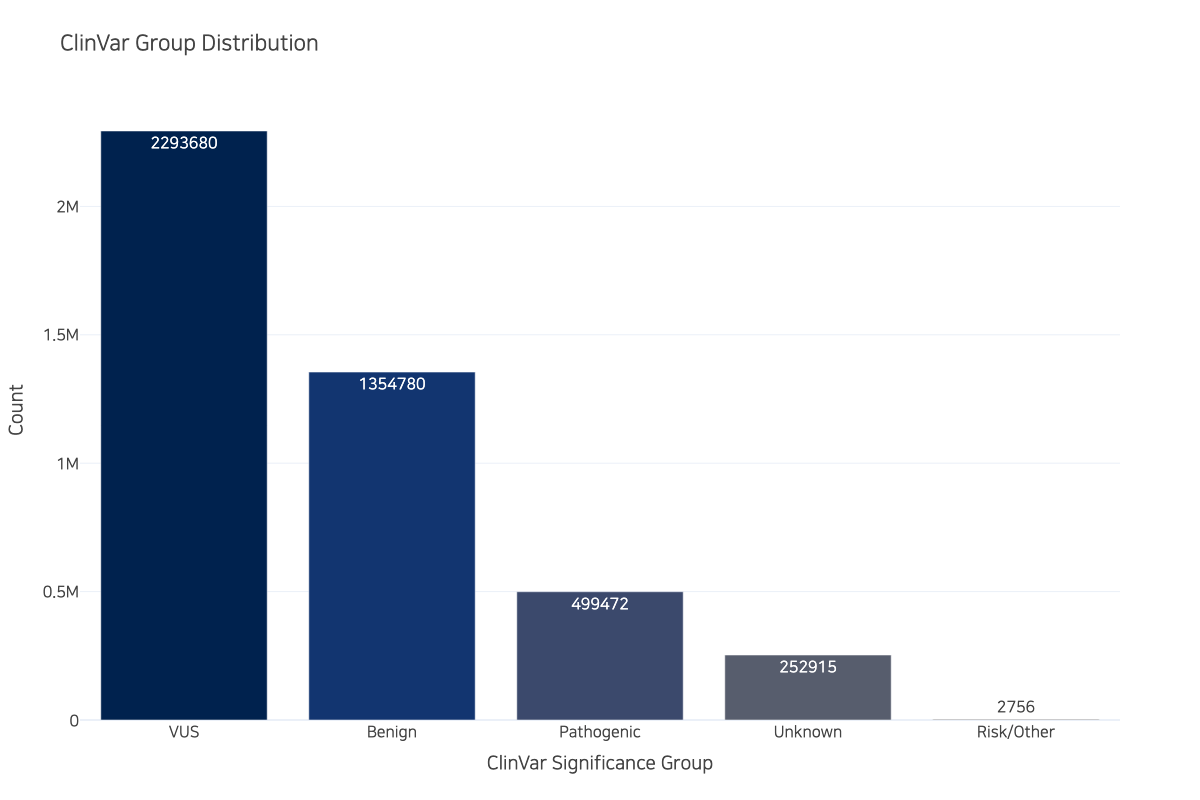
그 결과물이 이거임. 다행히도 폰트 전역으로 설정해둔 건 듣는다. 어 근데 저기 저장 코드는 왜 주석처리 했어요? Plotly는 그래프 복사가 안 되고 저장한 다음에 올려야 하는데, 문제는 그 저장하는걸 할 때 시간이 증말 완전 개같이 오래 걸립니다. 그래서 그래프를 보여줄때는 저 코드에 주석을 씌워놓고 그래프_최종_진짜최종_저장.png 느낌으로 저장할때만 주석을 해제하는 것이다. 참고로 저거 그냥 저장되는 거 아니고 kaleido 까셔야 합니다.
# 트리맵은 Plotly Express(px)가 훨씬 직관적입니다.
fig = px.treemap(
clnsig_grp,
path = ['CLNSIG_Group'], # 계층 구조 (여기선 그룹 하나)
values = 'Total',
color = 'Total', # 숫자에 따라 색상 농도 조절
color_continuous_scale = 'algae',
title = "ClinVar Significance Hierarchy"
)
# 전역 설정된 폰트 적용 확인
fig.update_layout(width=1200, height=800)
# fig.write_image('group_distributuion_heatmap.png')
fig.show()
이 트리맵에는 문제가 하나 있다. 저기 막대그래프를 보시면 Risk/Other가 다른 값들에 비해 비중이 좀 적죠? 예. 블록이 안보입니다. 착한 사람만 보이는 블록임.
fig = go.Figure()
fig.add_trace(go.Pie(
labels = clnsig_grp['CLNSIG_Group'],
values = clnsig_grp['Total'],
textinfo = 'label+percent', # 이름과 퍼센트 동시에 표시
insidetextorientation = 'radial',
marker = dict(colors=px.colors.sequential.algae) # 전역 설정 색감 유지
))
fig.update_layout(
title_text = "ClinVar Significance Proportion",
width = 1200,
height = 800,
margin = dict(t=50, l=10, r=10, b=10)
)
# fig.write_image('group_distributuion_pie.png')
fig.show()
여기서도 안보이죠? 이정도면 거의 주서터널현미경 빌려서 봐야됨… ㅋㅋ
CLNSIG-Chromosome
clnsig_grp = clinvar_df.group_by(['CLNSIG_Group', 'CHROM_Type']).agg(
pl.col('CLNSIG').count().alias("Total")
).sort('CLNSIG_Group')CLNSIG-염색체 그룹으로 묶어보았다. 언노운은 몇 번 염색체인지 불명인 것 같음.
max_total = clnsig_grp['Total'].max()
sizeref = 2. * max_total / (100**2) # 100은 최대 버블 반지름(픽셀)
fig = go.Figure()
# 2. 그룹별로 순회하며 트레이스 추가
# 이렇게 하면 전역 설정된 colorway 순서대로 색이 입혀집니다!
for group_name in clnsig_grp['CLNSIG_Group'].unique():
# 해당 그룹 데이터만 필터링
curr_df = clnsig_grp.filter(pl.col('CLNSIG_Group') == group_name)
fig.add_trace(go.Scatter(
x = curr_df['CLNSIG_Group'],
y = curr_df['CHROM_Type'],
name = group_name, # 범례에 표시될 이름
mode = 'markers+text',
marker = dict(
size = curr_df['Total'],
sizemode = 'area',
sizeref = sizeref,
sizemin = 10,
# 여기서 color를 지정하지 않으면 전역 설정(colorway)을 따라갑니다!
),
text = curr_df['Total'],
texttemplate = "%{x}<br>%{text:,d}",
textposition = "top right",
textfont = dict(family="NanumSquare_ac", size=12)
))
# 3. 레이아웃 설정 (컬러바는 어차피 안 나오지만 확인사살)
fig.update_layout(
width = 1200,
height = 800,
title = "ClinVar Significance by Group (Global Palette Applied)",
showlegend = True, # 그룹별로 색이 다르니 범례를 보여주는 게 좋습니다
)
# fig.write_image('cln-chr bubble.png')
fig.show()
스읍… 이거 색깔을 x축별로 바꾼건데… algae로 했더니 표가 너무 안남..

오… 일단 언노운 퇴근시키자.

굿. 저 버블이랑 라벨 간격은 우리가 조절을 못해요...
염색체별 CLNSIG 비율
fig = go.Figure()
fig.add_trace(
go.Bar(x = clnsig_grp['CHROM_Type'], y = clnsig_grp['Total'], marker_color = px.colors.sequential.algae, text = clnsig_grp['CLNSIG_Group'])
)
fig.update_layout(
width = 1200, height = 800,
xaxis=dict(title='Chromosome Type'),
yaxis=dict(title='Count'),
title = "ClinVar Group Distribution",
margin = dict(t=50, l=10, r=10, b=10)
)
# fig.write_image('group_distributuion.png')
fig.show()이거 일단 이대로도 볼 수는 있거든요? 근데 문제가 하나 있음.

악 내눈!
일단 왜 이런 사태가 일어났는지를 먼저 봐야 한다. clinVar 데이터에 들어있는 염색체가 언노운 빼고 총 25종인데 그 25종이 각개가 아니라 1~22번까지가 상염색체(남녀 다 똑같이 있는거), X랑 Y가 성염색체(남녀 분포 달라요), 그리고 미토콘드리아가 있어요. 이게 다 길이가 같다고 쳐도 쪽수때문에 일단 상염색체 물량이 압도적인데 염색체 그림 찾아보면 어때요? 1번 봐봐요. 크기가 벌써부터 이놈은 키가 커서 1번인가 하게 길어요…
그러니까 상염색체에 비하면 다른 염색체들은 거의 뭐 종합운동장에 축구공 하나씩 굴러다니는 정도로 미미한 수준이라 저렇게 나오는거다. 저걸 파훼할 방법이 있냐고?
# 1. 데이터에 있는 실제 값 순서 (가출 방지용)
real_values = ["Sex_Chrom", "Autosome", "Mitochondria"]
# 2. 그래프 위에 표시하고 싶은 예쁜 제목들 (순서 일치 필수!)
display_titles = ["Sex Chromosome", "Autosome", "Mitochondria"]
# 3. 서브플롯 생성
fig = make_subplots(
rows=1, cols=3,
subplot_titles=display_titles, # 여기서 예쁜 제목 적용!
horizontal_spacing=0.08
)
# 4. 반복문은 실제 값(real_values)으로 돌립니다
for i, real_val in enumerate(real_values):
curr_df = clnsig_grp.filter(pl.col('CHROM_Type') == real_val)
# 내림차순 정렬해서 보기 좋게
curr_df = curr_df.sort("Total", descending=True)
fig.add_trace(
go.Bar(
x = curr_df['CLNSIG_Group'],
y = curr_df['Total'],
text = curr_df['Total'],
texttemplate = '%{y:,}',
textposition = 'outside',
# 색깔은 이미 설정한 대로!
marker_color = px.colors.sequential.algae[-(i*3+1)]
),
row = 1, col = i + 1
)
# 5. 레이아웃 정리
fig.update_layout(
height=600,
width=1250,
title_text="ClinVar Distribution by Chromosome Type",
showlegend=False,
margin=dict(t=100, l=20, r=20, b=50), # 제목 공간 확보 위해 t(top) 늘림
)
# fig.write_image('group_distributuion_subplot.png')
fig.show()
서브플롯을 쓰던가
# 1. 비율(Percentage) 계산 (Polars 활용)
clnsig_perc = clnsig_grp.with_columns(
(pl.col("Total") / pl.col("Total").sum().over("CHROM_Type") * 100).alias("Percentage")
)
# 2. Plotly Express로 그리기 (비율 차트는 PX가 압도적으로 편합니다)
fig = px.bar(
clnsig_perc,
x = "CHROM_Type",
y = "Percentage",
color = "CLNSIG_Group", # 그룹별 색상
text = "Percentage",
title = "ClinVar Significance Proportion (100% Stacked)",
color_discrete_sequence = px.colors.qualitative.Vivid # 아까 쓰신 Prism!
)
fig.update_traces(texttemplate='%{text:.1f}%', textposition='inside')
fig.update_layout(width=1200, height=800, barmode='stack')
# fig.write_image('group_distributuion_stack.png')
fig.show()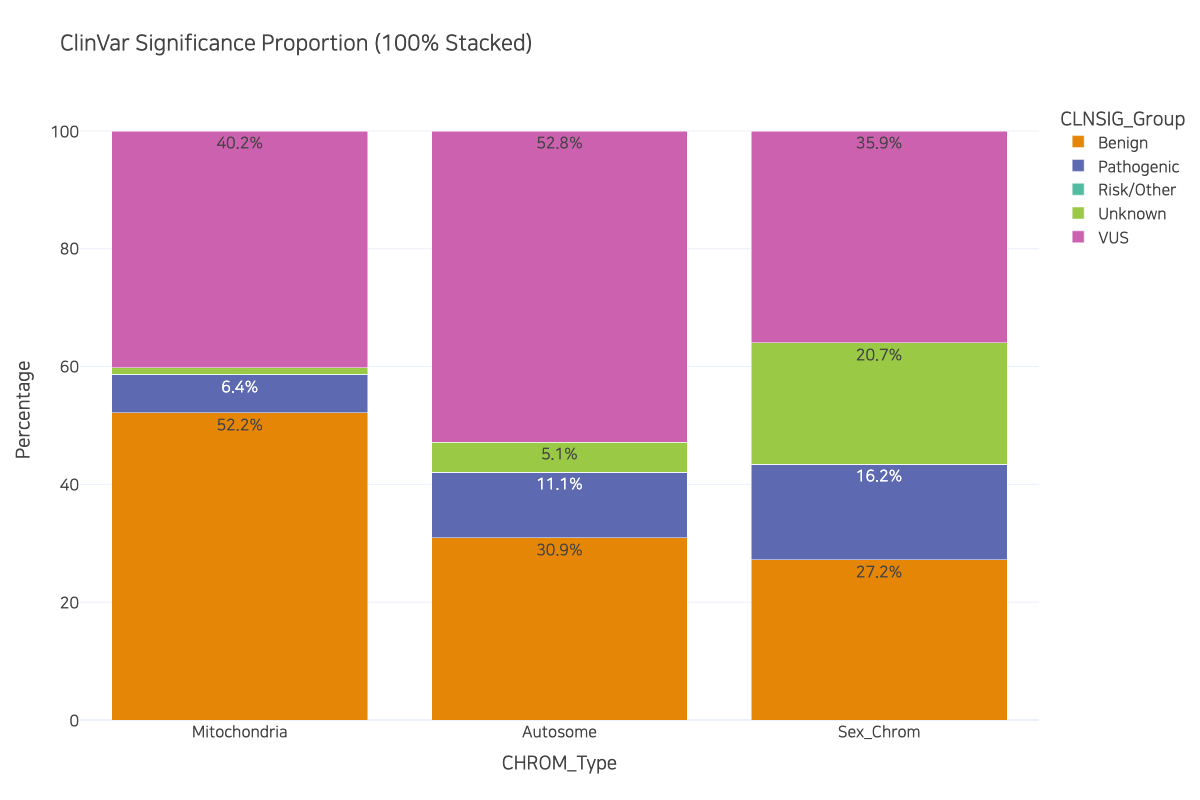
누적... 그리셔도 되는데 문제가 뭐다? Risk/Other가 안보여요...
이제 다음편에서 Pathogenic한 변이만 골라서 또 해볼거다. 이거 한번에 하면 님들 데이터 요금 급나나가요.
'Coding > EDA' 카테고리의 다른 글
| clinVar EDA를 Polars로 해보자-Appendix. 각 변이별 일짱 (0) | 2026.04.15 |
|---|---|
| clinVar EDA를 Polars로 해보자-Pathogenic EDA (0) | 2026.04.14 |
| Medical Cost Personal Datasets (0) | 2026.03.18 |
| Red Wine Quality (0) | 2026.03.13 |
| Palmer Archipelago (Antarctica) penguin data (0) | 2026.03.11 |
보통 판다스 데이터프레임 불러와서 지지고 볶고 뭐 해요? 그래프 그리죠. 표로 정리해서 보여주는것보다 그래프 딱 만들어서 도표 딱 보여주면 기깔나쟎아요? 그겁니다. 그리고 우리가 제일 많이 쓰는 맷플롭이나 씨본(+Plotly)에서도 폴라스를 받아줄지 궁금해서 해봤습니다.
이번에 써 볼 데이터프레임은 파일 불러온거 하나(켐플) 있고, 직접 만든거 하나 있습니다.
import polars as pl
import matplotlib.pyplot as plt
import seaborn as sns
import plotly.express as px # 프로젝트 이런거 아니니까 걍 얘 부를게여위에서부터 순서대로 폴라스, Matplotlib, 씨본, Plotly임다. 불러오십쇼. pyarrow는 불러올 필요 없고 설치만 하면 됨.
# 피보나치 수열
dict = {
'A':[1, 1, 2, 3, 5, 8, 13, 21, 34, 55],
'B':[1, 2, 3, 4, 5, 6, 7, 8, 9, 10]
}
# 을 데이터프레임으로
df = pl.DataFrame(dict)그리고 직접 만든 데이터는 이겁니다. 피보나치 수열임.
Matplotlib
둘다 잘 됨. 진짜로 그게 답니다. Null이 있으면 걍 이게 뭐여 패스 하는듯.


위가 피보나치, 아래가 산점도다. ...저기 0이 널 아니예요? 나도 그런 줄 알고 확인해봤는데 그냥 수소결합을 안 하는 놈임.
Seaborn


근데 왜 씨본은 색이 다르냐면 쟤는 Hue 옵션을 줬음. 쟤만 줬어요.
sns.scatterplot(load_df.to_pandas(), x = 'HBA', y = 'HBD')
plt.show()산점도는 그리다가 결측값때문에 에러가 났는데, 판다스는 NaN(날짜는 NaT)이지만 폴라스는 null이잖아요? 이걸 결측값 처리 안 하고 걍 주면 씨본이 이게 뭐시여 하고 오류를 토하니까 to_pandas()를 준 겁니다. 근데 저게 pyarrow가 설치되어있어야 가능하니까 또 설치한건데… 여러분들이 EDA를 할 거라면 안 쓸 칼럼은 날릴거고, 그게 아니어도 결측값을 어떻게든 채울거잖아요? 결측값 땜질만 적절하게 하면 오류는 볼 일이 없습니다.
Plotly

쟤는 왜 캡쳐했나요? 플롯틀리는 그래프 복사가 안됩니다. 쟤 사이즈 왜저래요? 사이즈가 불만이면 그릴때 조절해야 합니다. 조절 안하면 저렇게 넙대대한 놈 나옵니다.
아무튼 둘 다 오류 없이 잘 나온 건 맞다.
'Coding > Python' 카테고리의 다른 글
| Polars를 써보자 (0) | 2026.04.09 |
|---|---|
| MSA에 군집분석을 끼얹어보세요! (0) | 2026.02.20 |
| M1V1 = M2V2 (0) | 2026.02.15 |
| 코로나바이러스 MSA (0) | 2026.02.05 |
| 라이노바이러스 유전자로 MSA를 해보았다 (0) | 2026.01.27 |
엥? 폴라스? 그게 뭐예요오?
Polars is an open-source library for data manipulation, known for being one of the fastest data processing solutions on a single machine. It features a well-structured, typed API that is both expressive and easy to use.
뭐라는겨 싶겠지만 폴라스는 판다스 비슷한 일을 한다. 데이터프레임을 만들거나 불러오거나 하는 모든 일들이 가능한데 일단 읽는 속도가 판다스보다 20배 빨랐음. 뭐 메모리 어쩌고 하던데 나는 컴퓨터 아키텍쳐까지는 잘 모르니까 패스하고... 판다스랑 좀 다른 부분이 있다는 건 유념하십쇼.
import polars as pl일단 판다스처럼 폴라스도 깔고 불러야 쓸 수 있다. 넘파이...는... 불러야되나?
파~일을 열어다~오~
df_csv = pl.read_csv("data/Fluoxetine.csv", separator=";", null_values=["", '""', 'None']) # ChEMBL은 구분자가 세미콜론임다
df_csv2 = pd.read_csv("data/Fluoxetine.csv", sep=";")여기서 문제가 하나 있음. 저기서 어떤게 폴라스고 어떤게 판다스게요? 위에껍니다. 세부적인 옵션 빼고 걍 불러오는건 폴라스나 판다스나 비슷해서 pl pd로 구별해야되는데 이게 하다보면 그게 잘 안됩니다. 둘 중 하나만 쓰거나 둘 다 써야되는 상황이면 변수명으로 구별하던가 주석을 다십시오.
separator=";"는 csv 구분자 옵션인 거 알겠는데 저 뒤에놈은 뭔가요? 저게 켐블 불러온건데, 구분자가 ;이고 없는건 걍 없거든요? 그러면 판다스는 아 없군 결측값! 하는데 폴라스는 ""로 때워버립니다. 이게 그리고 NaN이랑 null이랑 달라요 폴라스는. 폴라스에서는 결측값이 널인데 이건 SQL의 널 비슷하게 흘러갑니다. ""는 어쨌든 뭐가 있는거니까 결측값이 아니래요. 그래서 부를때 자 이게 결측값이야~ 도 같이 해 준 거다.
참고로 같은 파일 불러오는데 폴라스는 2밀리초, 판다스는 21밀리초 걸림.
내 하드에 저-장
쓰는것도 코드 자체는 차이가 없다. to_csv 하면 되는'데' 저장한 결과물에서 차이가 난다. 판다스 써보신 분들 to_csv로 저장한거 다시 열었더니 맨 왼쪽 칼럼에 Unnamed:0 생긴 적 있으시죠? 그런거 한 두세번 돌리면 언네임드만 주루룩 생기고 그래서 열고 드롭한 적 있으시죠? 판다스에서 csv로 저장할때는 index=False를 줘야 그게 안 생긴다.
그럼 폴라스는요? 폴라스는 그 옵션 안 줘도 인덱스가 따로 저장이 안 된다. 대신 저장하는데 드는 소요시간은 비슷했음.
결측값
위에도 썼지만 결측값이 폴라스는 널이다. 그래서 .isna() 비슷한 함수가 .is_null()이고 .isna().sum() 비슷한건 .null_count()인데... 이거 가로로 나옵니다... 그리고 폴라스는 표 출력하면 위에 shape가 같이 나옴.
쿼리 되나요?
# 1. 컬럼명 변경 (성공적)
df_csv = df_csv.rename({"ChEMBL ID": "ChEMBL_ID"})
# 2. 컨텍스트 및 테이블 등록
ctx = pl.SQLContext()
ctx.register("chembl_table", df_csv)
# 3. 쿼리 실행 (쌍따옴표 제거!)
result = ctx.execute("""
SELECT * FROM chembl_table
WHERE ChEMBL_ID = 'CHEMBL153036'
""").collect()
result폴라스에서는 당신이 SQL 쌉고인물이라면 파이썬으로 불러와놓고 SQL 쿼리를 쓸 수 있다. 실화임. SELECT * FROM chembl_table WHERE ChEMBL_ID = 'CHEMBL153036' <<이게 SQL 쿼리예요.
df_csv.filter(pl.col("ChEMBL ID") == "CHEMBL153036")아니 그럼 SQL 알못들은 어쩌라고요! 아 필터 쓰십쇼.
데이터의 정보를 확인하는 방법
요즘 EDA할때 생략하는 그거 맞다. 폴라스... 하게되면 이거는 정보 확인하는거 한번 올려드림. 판다스랑 기능은 똑같은데 토해내는게 달라요.
df_csv.schema.info() 비슷한 기능을 하는게 .schema인데 인포처럼 칼럼 몇갠데 몇개 비었고 이런건 안나오고 뭔 타입인지만 나온다. 인포 비슷한거 필요하시면 .glimpse() 쓰십쇼. 아니면 널카운트?
아니면 .describe()에서 칼럼별 통계값 보고 알 수 있다. 예? 뭔 소리임? 폴라스에서는 널이 결측값이라고 했잖아요? 근데 SQL에서 연산에 널끼면 다 널됩니다. 왜냐고요? 널이 응 아니 몰라에서 몰라거든. 설문조사로 치자면 예 아니오 무응답에서 무응답이다. 슈뢰딩거의 고양이는 박스 까보면 얘가 갔는지 있는지는 알 수 있지만 널은 박스 밀봉된 슈뢰딩거의 고양이라 진짜 모름.
이 글에서는 그냥 데이터 열고 정보 확인하는것만 비교해봤는데, 다음편에서는 시각화 어떻게 되나 해 볼 예정이다. matplotlib나 seaborn에서는 폴라스를 못 쓴다는 얘기가 있음.
'Coding > Python' 카테고리의 다른 글
| Polars 데이터프레임도 시각화가 되나요? (0) | 2026.04.10 |
|---|---|
| MSA에 군집분석을 끼얹어보세요! (0) | 2026.02.20 |
| M1V1 = M2V2 (0) | 2026.02.15 |
| 코로나바이러스 MSA (0) | 2026.02.05 |
| 라이노바이러스 유전자로 MSA를 해보았다 (0) | 2026.01.27 |
이거어어어어는... 돌리는건 하나 돌렸습니다. 그럼 왜함? 그 옵튜나인가 뭔가 하는 거 써보려고 했음.
Optuna
파이썬 라이브러리인데, 하이퍼파라미터 튜닝할때 쓴다. 모델 학습 전 사용자가 직접 설정하는 외부 구성 변수를 하이퍼파라미터라고 하는데, 이걸 사람이 수동으로 일일이 조정해가면서 어느 조건에서 내 모델을 뽕을 잘 뽑을지를 고민...하면서 일일이 하다 보면 할 것도 많고 파라미터 하나하나 일일이 손대가면서 찾기도 힘들잖아요? 그 노가다를 알아서 해주는게 옵튜나입니다.
모델 바이 모델이라 함수에 집어넣는 게 다른데, 아무튼 이거 이거 이거 해줘 하면 지 알아서 음 이렇군 하면서 오케이 가릿 이걸로 진행시켜 한다. 모델에 따라서는 시간이 좀 걸리기도 합니다.
안하면 섭한 전처리
자녀 수 범주화
# 자녀 수는 모르겠고 유무로 범주화할거임. ㅇㅋ? ㅇㅇㅋ.
cost['have_child'] = cost['children'].apply(lambda x: 0 if x == 0 else 1) # 자녀수가 0이면 0, 0보다 크면 1
# 유자녀가 1입니다심플하게 유자녀 무자녀로 범주화했다.
일부 칼럼 퇴근시키기
cost.drop('region', axis = 1, inplace = True)cost.drop('children', axis = 1, inplace = True)자녀 칼럼: 범주화했으니까 퇴근해도 됨/지역 칼럼: 학습에 도움 안 될 것 같아서 퇴근
스케일뤄어어어어
scaler = StandardScaler()
cost[['age','bmi']] = scaler.fit_transform(cost[['age','bmi']])이정도면 다들 저거 뭔지 아시죠?
매핑
# 흡연여부
cost['smoker'] = cost['smoker'].map({'yes': 1, 'no': 0})cost['sex'] = cost['sex'].map({'male': 1, 'female': 0})아니 성별은 원핫인코더 쓰려고 했더니 에러토하데… ㅡㅡ
옵튜나 써보기
# 1. 독립변수(X)와 종속변수(y) 분리
X = cost.drop('charges', axis=1)
y = np.log1p(cost['charges']) # 아까 말씀드린 로그 변환!
# 2. 데이터 분할 (보통 8:2나 7.5:2.5로 나눕니다)
X_train, X_test, y_train, y_test = train_test_split(
X, y, test_size=0.2, random_state=42
)
print(f"학습 데이터 개수: {len(X_train)}")
print(f"테스트 데이터 개수: {len(X_test)}")근데 우리 이거 쓰기 전에 학습용 테스트용 나눠야되는건 아시죠?
def objective(trial):
param = {
'n_estimators': trial.suggest_int('n_estimators', 500, 2000),
'max_depth': trial.suggest_int('max_depth', 3, 9),
'learning_rate': trial.suggest_float('learning_rate', 0.01, 0.1),
'subsample': trial.suggest_float('subsample', 0.6, 1.0),
'colsample_bytree': trial.suggest_float('colsample_bytree', 0.6, 1.0),
'random_state': 42
}
model = XGBRegressor(**param)
model.fit(X_train, y_train)
preds = model.predict(X_test)
rmse = np.sqrt(mean_squared_error(y_test, preds))
return rmse
# 최적화 시작
study = optuna.create_study(direction='minimize')
study.optimize(objective, n_trials=50) # 50번 정도만 돌려봐도 감이 옵니다.
print(f"최고의 파라미터: {study.best_params}")이렇게 범위를 정해주면... 그 범위 뭐가 뭔지 모르겠으면 에미나이나 지피티, 클로드한테 나 이 모델 돌릴건데 옵튜나 짜줘 하시면 됩니다... 아무튼. 저렇게 주고 맞춰주십쇼 하면 옵튜나가 아 ㅇㅋㅇㅋ 해봅시다! 하면서 뭔가 돌아가요.
최고의 파라미터: {'n_estimators': 815, 'max_depth': 3, 'learning_rate': 0.017046309566345508, 'subsample': 0.9510680208670862, 'colsample_bytree': 0.8874051559739393}그리고 다 돌아가면 이렇게 돌리면 이렇게 나온다! 하고 알려줍니다. 회귀 돌릴때 옵튜나 쓰면 최상의 파라미터랑 그걸로 돌렸을때 R^2도 같이 나옴.
학습
model = XGBRegressor(**study.best_params)
model.fit(X_train, y_train)하아니 그럼 저 파라미터를 다 복붙해야 하나요? 아니, 그런 귀찮은 짓을 할 필요가 없어요. 우리는 저 파라미터를 변수에 할당해뒀으니까 그거 앞에 애스터리스크 두개 붙여서 모델에 넣으십쇼.
# 1. 예측 수행 (로그 상태)
y_pred_log = model.predict(X_test)
# 2. 역변환 (로그 -> 원래 달러 단위)
y_pred = np.expm1(y_pred_log)
y_actual = np.expm1(y_test)
# 3. 성능 지표 확인
print(f"R² Score (결정계수): {r2_score(y_actual, y_pred):.4f}")
print(f"MAE (평균 절대 오차): ${mean_absolute_error(y_actual, y_pred):.2f}")
print(f"MSE (평균 제곱 오차): ${mean_squared_error(y_actual, y_pred):.2f}")
print(f"RMSE (제곱근 평균 제곱 오차): ${np.sqrt(mean_squared_error(y_actual, y_pred)):.2f}")R² Score (결정계수): 0.8768
MAE (평균 절대 오차): $1999.14
MSE (평균 제곱 오차): $19125564.44
RMSE (제곱근 평균 제곱 오차): $4373.28?? 근데 내 예상보다 더 잘나왔어 이거...
'Coding > EDA' 카테고리의 다른 글
| clinVar EDA를 Polars로 해보자-Pathogenic EDA (0) | 2026.04.14 |
|---|---|
| clinVar EDA를 Polars로 해보자 (0) | 2026.04.13 |
| Red Wine Quality (0) | 2026.03.13 |
| Palmer Archipelago (Antarctica) penguin data (0) | 2026.03.11 |
| Google Play Store – Most Downloaded Android Apps (0) | 2026.03.02 |
오늘은 회귀입니다. …회기는 그 경희대 있는 지하철역이예요 선생님들.
주성분분석
머고 오늘은 ML 안함? 아니 할건데… 저게 회귀분석 할거라고 했죠? 저 데이터 칼럼이 12개인데 하나빼고 다 독립변수니까 지금 독립변수가 11개거든요. 그거 그대로 때려박으면 다중공선성 터질수도 있으니까 압축할 수 있는건 압축하고 가자 이겁니다. 11개 언제 넣었다 뺐다 할거임?
wine_x = wine.copy()
X = wine_x.drop('quality', axis=1)
y = wine['quality']
# 1. 스케일링 (PCA 전 필수!)
scaler = StandardScaler()
X_scaled = scaler.fit_transform(X)
# 2. PCA 객체 생성 (일단 모든 성분을 다 뽑아봅니다)
pca = PCA()
X_pca = pca.fit_transform(X_scaled)그나마 데이터가 다 수치형이라 FAMD 안 가고 주성분분석 했음.
# 전체 성분(11개)에 대해 PCA 수행
pca_full = PCA().fit(X_scaled)
# 개별 분산 설명력
individual_var = pca_full.explained_variance_ratio_
# 누적 분산 설명력
cum_var = np.cumsum(individual_var)
# 결과 출력
for i, (ind, cum) in enumerate(zip(individual_var, cum_var)):
print(f"PC{i+1}: 개별 {ind:.2f} / 누적 {cum:.2f}")
if cum >= 0.85 and i > 0 and cum_var[i-1] < 0.85:
print(f"--- 여기까지 딱 끊으면 정보의 {cum*100:.1f}%가 보존됩니다! ---")PC1: 개별 0.28 / 누적 0.28
PC2: 개별 0.18 / 누적 0.46
PC3: 개별 0.14 / 누적 0.60
PC4: 개별 0.11 / 누적 0.71
PC5: 개별 0.09 / 누적 0.80
PC6: 개별 0.06 / 누적 0.86
--- 여기까지 딱 끊으면 정보의 85.5%가 보존됩니다! ---
PC7: 개별 0.05 / 누적 0.91
PC8: 개별 0.04 / 누적 0.95
PC9: 개별 0.03 / 누적 0.98
PC10: 개별 0.02 / 누적 0.99
PC11: 개별 0.01 / 누적 1.00제 6 주성분까지 채용하면 85% 이상의 설명력을 갖는다. 그래서 그 주성분들이 뭔데?
for i in range(n_comp):
loading_scores = pd.Series(
pca_full.components_[i],
index=X.columns
)
sorted_loadings = loading_scores.abs().sort_values(ascending=False)
print(f"\nPC{i+1} 핵심 변수 TOP3")
print(sorted_loadings.head(3))PC1 핵심 변수 TOP3
fixed acidity 0.489314
citric acid 0.463632
pH 0.438520
dtype: float64
PC2 핵심 변수 TOP3
total sulfur dioxide 0.569487
free sulfur dioxide 0.513567
alcohol 0.386181
dtype: float64
PC3 핵심 변수 TOP3
alcohol 0.471673
volatile acidity 0.449963
free sulfur dioxide 0.428793
dtype: float64
PC4 핵심 변수 TOP3
chlorides 0.666195
sulphates 0.550872
residual sugar 0.372793
dtype: float64
PC5 핵심 변수 TOP3
residual sugar 0.732144
alcohol 0.350681
pH 0.267530
dtype: float64
PC6 핵심 변수 TOP3
pH 0.522116
volatile acidity 0.411449
density 0.391152
dtype: float64이 에미나이 변수 압축한다고 했는데 산점도 볼때부터 알아봤어야 했는데…
1. 제 1주성분: fixed acidity, citric acid, pH
2. 제 2주성분: total sulfur dioxide, free sulfur dioxide, alcohol
3. 제 3주성분: alcohol, volatile acidity, free sulfur dioxide
4. 제 4주성분: chlorides, sulphates, residual sugar
5. 제 5주성분: residual sugar, alcohol, pH
6. 제 6주성분: pH, volatile acidity, density
이렇게 나왔으면 중복 쳐내면 됩니다.
{'free sulfur dioxide', 'fixed acidity', 'sulphates', 'pH', 'total sulfur dioxide', 'residual sugar', 'density', 'chlorides', 'alcohol', 'volatile acidity', 'citric acid'}저거 리스트에 다 때려박고 set으로 바꾸면 중복 다 쳐냄.
회귀분서억
X = X_scaled # 저기 주성분 돌리기 전에 거쳤어요 스케일러
model = LinearRegression()
model.fit(X, y)
pred = model.predict(X)print("MAE:", mean_absolute_error(y, pred))
print("MSE:", mean_squared_error(y, pred))
rmse = np.sqrt(mean_squared_error(y, pred))
print("RMSE:", rmse)
print("R²:", r2_score(y, pred))MAE: 0.5004899635644883
MSE: 0.416767167221408
RMSE: 0.6455750670692045
R²: 0.3605517030386882거 설명력이 너무 약한 거 아니오?

잔차분석 했더니 고질라 왔다간거 실화냐?
VIF(다중공선성)
이거 왜 보냐면 독립변수끼리 상관이 있나를 보는겁니다. 지들끼리 상관이 있으면 모델 시망됨.
wine_x = wine_x.drop('quality', axis=1)
# X는 독립변수 데이터프레임 (스케일링 안 해도 되지만 보통 해도 상관없음)
X_vif = pd.DataFrame()
X_vif["variable"] = wine_x.columns
X_vif["VIF"] = [variance_inflation_factor(wine_x.values, i) for i in range(wine_x.shape[1])]
print(X_vif.sort_values(by="VIF", ascending=False)) variable VIF
7 density 1479.287209
8 pH 1070.967685
10 alcohol 124.394866
0 fixed acidity 74.452265
9 sulphates 21.590621
1 volatile acidity 17.060026
2 citric acid 9.183495
4 chlorides 6.554877
6 total sulfur dioxide 6.519699
5 free sulfur dioxide 6.442682
3 residual sugar 4.662992망했는데…? 들어내봐야겠지 이거…?
부록-알콜과 퀄리티
wine.corr()['quality'].sort_values(ascending=False)quality 1.000000
alcohol 0.476166
sulphates 0.251397
citric acid 0.226373
fixed acidity 0.124052
residual sugar 0.013732
free sulfur dioxide -0.050656
pH -0.057731
chlorides -0.128907
density -0.174919
total sulfur dioxide -0.185100
volatile acidity -0.390558
Name: quality, dtype: float64알콜이 상관계수가 제일 높은데?
plt.figure()
scatter = plt.scatter(
wine['alcohol'],
wine['quality'],
c=wine['quality'],
cmap='viridis',
alpha=0.6
)
plt.xlabel('Alcohol')
plt.ylabel('Quality')
plt.title('Alcohol vs Wine Quality')
plt.colorbar(scatter, label='Quality')
plt.show()

이냥반들아 알콜 많이 들어가봐야 소독용 에탄올이라고... 내가 그래서 고량주 안마심. 실험실에서 맡던 소독용 에탄올 냄새 나서...
부록 2-XGBoost가 여기서 왜 나와요
우리 방금까지 회귀했는데 쟈는 또 뭐임? 어제 그 분류했던 친구입니다. 이 데이터셋으로 회귀도 하고 분류도 한다고 함.
# 일단 쨈
wine_df = wine.copy()
X = wine_df.drop("quality", axis=1)
y = wine_df["quality"]X_train, X_test, y_train, y_test = train_test_split(
X, y, test_size=0.2, random_state=42
)
scaler = StandardScaler()
X_train_scaled = scaler.fit_transform(X_train)
X_test_scaled = scaler.transform(X_test)model = XGBRegressor(
n_estimators=300,
max_depth=4,
learning_rate=0.05,
random_state=42
)
model.fit(X_train, y_train)
pred = model.predict(X_test)
print("R2:", r2_score(y_test, pred)) # R2: 0.4512701630592346어째 회귀보다 분류가 설명력이 더 좋은 것 같다.
SHAP
얘는 또 뭥미? 이건 그러니까 XGBoost와 대화의 시간을 가지면서 왜 그렇게 분류한건지 물어보는 친구다. 오은영박사님 어… 아니 생각하는 의자까지는 아니고…
# 1. 모델의 predict 함수를 직접 전달
# 2. masker를 사용하여 데이터의 통계적 분포를 SHAP에게 알려줍니다.
masker = shap.maskers.Independent(data=X_test)
explainer = shap.Explainer(xgb_model.predict, masker)
# 3. SHAP 값 계산 (Permutation 방식은 속도는 좀 걸리지만 매우 정확합니다)
shap_values = explainer(X_test)
# 4. 시각화
shap.summary_plot(shap_values, X_test)
그니까 이제 XGBoost한테 저 샤프가 오은영박사님에 빙의해서 왜 이렇게 분류했냐고 물어봤을 거 아닙니까? 그러니까 모델이 인제 얘를 이렇게 분류한 이유를 얘기해주는겁니다. 얘는 산도가 너무 높아서 이렇게 했고 얘는 밀도가 이래서 이렇게 분류했어요, 이렇게. 그게 저 그래프임다.
'Coding > EDA' 카테고리의 다른 글
| clinVar EDA를 Polars로 해보자 (0) | 2026.04.13 |
|---|---|
| Medical Cost Personal Datasets (0) | 2026.03.18 |
| Palmer Archipelago (Antarctica) penguin data (0) | 2026.03.11 |
| Google Play Store – Most Downloaded Android Apps (0) | 2026.03.02 |
| 얘! clinvar도 EDA가 된단다! (3) (0) | 2026.02.18 |
예… 그… 펭귄 데이터입니다. 그건 아는데 이걸 왜 꺼냈냐… 분류 할거라서 꺼냈습니다. 예. 아니 진짜 할거임.
전처리 하기 전에...
그냥 EDA였으면 범주화하고 결측값 확인하고 채우거나 날리거나 했을텐데, 이번에는 그렇게 하고 땡 하면 안된다. 왜냐… 무작정 아무 칼럼이나 학습하는데 썼다간 모델 성능이 떨어지거든요. 그리고 범주형 칼럼중에 학습에 쓸 칼럼은 인코딩도 해 줘야 한다.
학습에 쓸 수 없는 칼럼 날리기
내 일일이 올리기 귀찮아서 올리지는 않는다만... 분석하기 전에 항상 .head()랑 .column 써서 뭐 있는지 보고 가죠? shape는 잘 안씀... 아무튼. 거기서 칼럼들을 확인해보고 우선 날릴 것부터 정할거다.
# Copy
penguin_drop = penguin.copy()
# 하고 날려날려 칼럼
penguin_drop.drop(['studyName','Sample Number','Region','Individual ID','Clutch Completion', 'Comments', 'Date Egg', 'Stage'], axis=1, inplace=True)
penguin_drop.head()이걸 날리는 이유는 되게 간단하다. 학습에 도움이 안 돼서.
결측값 채우기
sns.kdeplot(penguin_drop['Culmen Length (mm)'])
롸? 갑자기 쟤가 왜 나옴? 임퓨터가 평균, 중앙값, 최빈값으로 채워주는건데 문제가 하나 있습니다. 그게 값 분포에 따라 적절한 걸 골라야지 덮어놓고 히히 평균해야징 하다간 모델 성능이 똥멍청이 1이 됩니다. 그래서 때우기 전에 분포를 본 건데, 이게 보니까… 저거 그 어린왕자 그 코끼리 그거 아님? 그럼 어떻게 합니까?
num_cols = ['Culmen Length (mm)', 'Culmen Depth (mm)', 'Flipper Length (mm)', 'Body Mass (g)'] # 일단 떄울 칼럼
imputer = KNNImputer(n_neighbors=5) # 얘는 그 이웃 참고해서 때워주는 친구입니다
penguin_drop[num_cols] = imputer.fit_transform(penguin_drop[num_cols]) # 때-움임퓨터중에 KNN Imputer라는 게 있는데, 얘는 결측값을 이웃한 그룹들을 참고해서 채워주는 친구다. 그렇게 해서 몸무게까지 네개 채워주면 일단 끝… 왜 저걸 일괄로 채움? 이유는 간단하다. 저 칼럼에 결측값이 두개씩 있었는데 그 두개가 같은 펭귄에서 빠져있었거든…
# . 대치합니닷
penguin_drop['Sex'] = penguin_drop['Sex'].replace('.', np.nan)
penguin_drop['Sex'].unique()성별은 범주형이라 최빈값으로 채워야 하는데, 그거 말고도 다른 문제가 있다. 저 점 뭐야 점. 저것도 결측값으로 바꾸고 최빈값으로 때울거다.
imputer = SimpleImputer(strategy='most_frequent')
penguin_drop[['Sex']] = imputer.fit_transform(penguin_drop[['Sex']]) # 때-움됐으. 이제 동위원소 때우러 가자.
num_cols = ['Delta 15 N (o/oo)', 'Delta 13 C (o/oo)'] # 일단 떄울 칼럼
imputer = KNNImputer(n_neighbors=5) # 얘는 그 이웃 참고해서 때워주는 친구입니다
penguin_drop[num_cols] = imputer.fit_transform(penguin_drop[num_cols]) # 때-움이것도 걍 KNN으로 때웠고, 이제 남은건 범주형 변수 인코딩하고 수치형 변수 스케일러 적용하는거다.
스탠다드 스케일러
scaler = StandardScaler() # 스케일러가 요기잉눼?
num_features = ['Culmen Length (mm)', 'Culmen Depth (mm)', 'Flipper Length (mm)',
'Body Mass (g)', 'Delta 15 N (o/oo)', 'Delta 13 C (o/oo)'] # 스케일러
cat_features = ['Island', 'Sex'] # 인코더
preprocessor = ColumnTransformer(
transformers=[
('num', StandardScaler(), num_features),
('cat', OneHotEncoder(handle_unknown='ignore'), cat_features)
])
X_processed = preprocessor.fit_transform(penguin_drop)일단 저 칼럼 트랜스포머가 뭐 하는 친구인지는 모르겠고 저게 다 된 거 맞습니다. 이대로 학습 들어가면 되는'데'...
이제 학습해야징 히히
y = penguin_drop['Species'] # 문제지
X_train, X_test, y_train, y_test = train_test_split(X_processed, y, test_size = 0.2, random_state=42, stratify=y) # 기본값 8:2
print(f'전체 데이터의 수 : {len(X_processed)}')
print(f'학습 데이터의 수 : {len(X_train)}')
print(f'테스트 테이터의 수 : {len(X_test)}')있어봐요 우리 모델도 아직 못골랐어... 일단 나눈거야... 저거 참고로 비율은 보통 5:3:2가 국룰인데, 5는 학습용이고 3은 과적합 여부 확인용, 2가 테스트용이다.
랜덤포레스트
forest = RandomForestClassifier(random_state=42) # 얘는 근데 숲이랑 뭔 상관이 있길래 이름이 랜덤포리스트인겨
forest.fit(X_train, y_train) # 학습
forest_pred = forest.predict(X_test)이게 다냐고? 예. 구글링했더니 뭐가 막 장황하게 나오긴 했는데 기본적으로는 이게 다다.
# 얼마나 맞췄는지 점수(%) 확인
print(f"정확도: {accuracy_score(y_test, forest_pred):.3f}")
# 종별로 얼마나 잘 분류했는지 상세 리포트
print(classification_report(y_test, forest_pred))정확도: 0.986
precision recall f1-score support
Adelie Penguin (Pygoscelis adeliae) 0.97 1.00 0.98 30
Chinstrap penguin (Pygoscelis antarctica) 1.00 0.93 0.96 14
Gentoo penguin (Pygoscelis papua) 1.00 1.00 1.00 25
accuracy 0.99 69
macro avg 0.99 0.98 0.98 69
weighted avg 0.99 0.99 0.99 69????? 아니 이게 내가 예상했던것보다 너무 잘나왔는데? 나 한 80퍼 되나 했는데 이게 뭐시여?????? 되게 지금 당황스러운데? 근데 아델리펭귄에 대한 정밀도는 좀 떨어지는데, 이게 그 결측값때문일수도 있다.

이 모델이 펭귄을 분류하는 데 있어서 중요하게 생각한 게 뭘까? 에 대한 답. 부리 길이와 날개 길이, 그리고 13-탄소가 피쳐 TOP 3인데... 저 동위원소 뭔데요? 저거 먹이활동같은거 추적할때 넣는 방사성 동위원소입니다.
SVM(서포트 벡터 머신)
svm_model = SVC(kernel='rbf', C=1.0, random_state=42)
svm_model.fit(X_train, y_train)
svm_predictions = svm_model.predict(X_test)# 1. 성적표 (Classification Report)
print("--- SVM 분류 성적표 ---")
print(classification_report(y_test, svm_predictions))
# 2. 오답 분석 (Confusion Matrix)
cm = confusion_matrix(y_test, svm_predictions)
disp = ConfusionMatrixDisplay(confusion_matrix=cm, display_labels=svm_model.classes_)
disp.plot(cmap='viridis')
plt.title("SVM Confusion Matrix")
plt.show()--- SVM 분류 성적표 ---
precision recall f1-score support
Adelie Penguin (Pygoscelis adeliae) 1.00 1.00 1.00 30
Chinstrap penguin (Pygoscelis antarctica) 1.00 1.00 1.00 14
Gentoo penguin (Pygoscelis papua) 1.00 1.00 1.00 25
accuracy 1.00 69
macro avg 1.00 1.00 1.00 69
weighted avg 1.00 1.00 1.00 69어… 이게… 이렇게 됨?
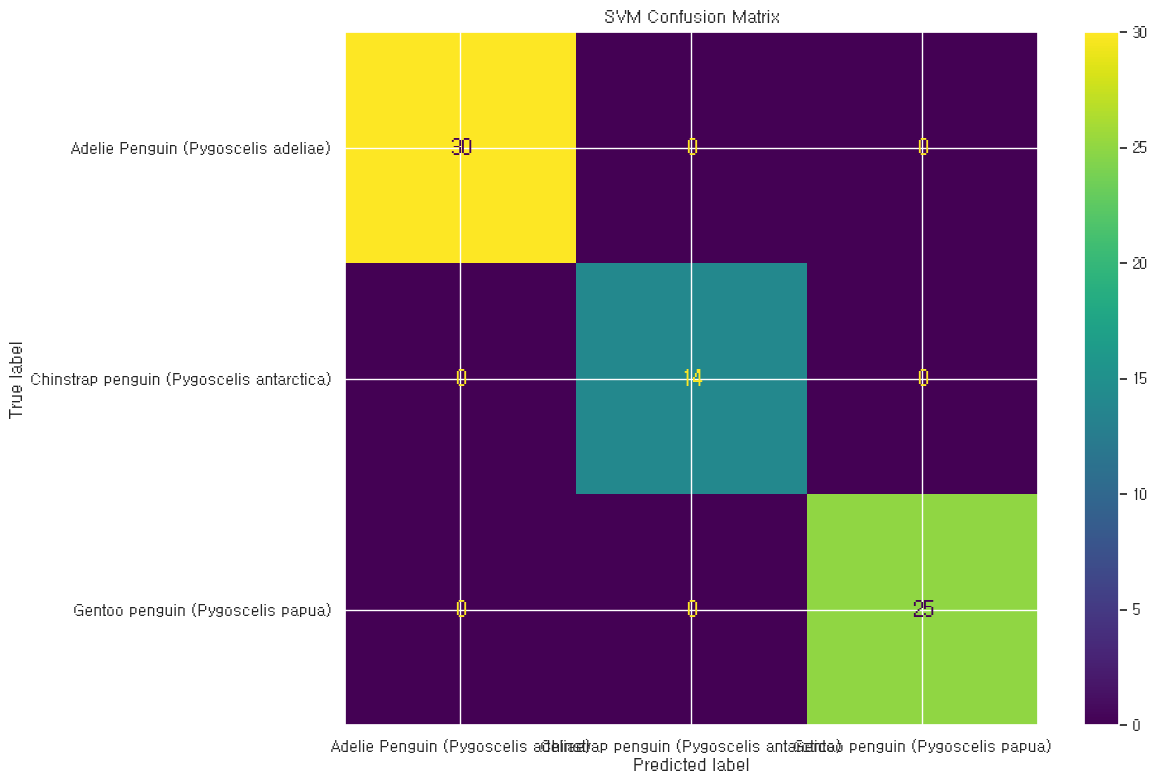
이거는… 걍 히트맵 그리는게 나을듯… 저 선 진짜 거슬려요.
XGBoost
# 1. 변환기 생성
le = LabelEncoder()
# 2. 정답지(y)를 숫자로 변환
y_train_encoded = le.fit_transform(y_train)
y_test_encoded = le.transform(y_test)
# 다중 분류이므로 objective를 'multi:softmax'로 설정하는 게 정석입니다
xgb_model = XGBClassifier(objective='multi:softmax', n_estimators=50, random_state=42)
xgb_model.fit(X_train, y_train_encoded)
# 결과 확인
print(classification_report(y_test, le.inverse_transform(xgb_model.predict(X_test))))롸? 라벨인코더 쟤는 왜 나옴? 쟤는 답지도 인코딩해줘야 학습합니다... 그리고 XGBoost랑 LightGBM은 싸이킷런에 없으니까 따로 설치하십쇼.
precision recall f1-score support
Adelie Penguin (Pygoscelis adeliae) 0.97 1.00 0.98 30
Chinstrap penguin (Pygoscelis antarctica) 1.00 0.93 0.96 14
Gentoo penguin (Pygoscelis papua) 1.00 1.00 1.00 25
accuracy 0.99 69
macro avg 0.99 0.98 0.98 69
weighted avg 0.99 0.99 0.99 69랜덤포레스트랑 비슷한디…?
LightGBM
얘도 설치하셔야됩니다… lightbgm 쳐놓고 왜 못깔지 이러고 있었음…ㅋㅋㅋㅋ
lgbm_model = lgb.LGBMClassifier(n_estimators=10, random_state=42, verbose=-1)
lgbm_model.fit(X_train, y_train_encoded)
# 1. 모델이 예측한 값(숫자)을 받아옵니다
lgb_pred_encoded = lgbm_model.predict(X_test)
# 2. 숫자를 다시 원래 펭귄 이름(문자열)으로 돌립니다
# 여기서 le(LabelEncoder)가 아까 학습(fit)된 상태여야 합니다
lgb_pred = le.inverse_transform(lgb_pred_encoded)쟤는 좀 적죠…? 50번 돌렸더니 10번만에 여까지해 됐어 퍼펙트해 하고 모델이 GG쳤음…
print("--- LightGBM 분류 성적표 ---")
print(classification_report(y_test, lgb_pred))
# 3. 마지막 혼동 행렬 시각화
cm = confusion_matrix(y_test, lgb_pred)
disp = ConfusionMatrixDisplay(confusion_matrix=cm, display_labels=lgbm_model.classes_)
disp.plot(cmap='Greens')
plt.title("LightGBM Confusion Matrix")
plt.show()--- LightGBM 분류 성적표 ---
precision recall f1-score support
Adelie Penguin (Pygoscelis adeliae) 0.94 0.97 0.95 30
Chinstrap penguin (Pygoscelis antarctica) 1.00 0.86 0.92 14
Gentoo penguin (Pygoscelis papua) 0.96 1.00 0.98 25
accuracy 0.96 69
macro avg 0.97 0.94 0.95 69
weighted avg 0.96 0.96 0.96 69
빨리 GG친 것 치고는 니가 성적이 제일 꼴찌여 이자식아...
근데 그럴수밖에 없는게 XGBoost나 LightGBM 둘 다 스케일이 큰 애들에 특화되어 있습니다. 그 미국에서 호미 엄청 사가는거 아십니까? 거기는 막 옥수수밭에서 길 잃어먹는 사람도 있을 정도로 밭이 커갖고 농기구들도 스케일이 장난 아니예요. 근데 그게 호미랑 뭔 상관이냐고? 그 농기구들로 정원 손질하려니 세밀하게 안되는거지 이제. 잡초 뽑으려다가 엄한 꽃도 다치고 그러거든요. 근데 호미는 미쿡 농기구 스케일에 비하면 스케일도 작지, 하나로 다 하지… 그거임다.
아래 두 개는 입장에서 펭귄 데이터갖고 분류하라는건 미쿡 농기구로 화분에 꽃심는격임.
'Coding > EDA' 카테고리의 다른 글
| Medical Cost Personal Datasets (0) | 2026.03.18 |
|---|---|
| Red Wine Quality (0) | 2026.03.13 |
| Google Play Store – Most Downloaded Android Apps (0) | 2026.03.02 |
| 얘! clinvar도 EDA가 된단다! (3) (0) | 2026.02.18 |
| 얘! clinvar도 EDA가 된단다! (2) (0) | 2026.02.17 |
처음에는 이거 또 복잡한거 아냐? 했는데 진짜 간단합니다.
1. 깃헙에 저장소를 만들 때 저장소 이름을 (프로젝트명).github.io로 한다.
2. 그 폴더에 readme.md 생성된걸 지우고 HTML, CSS, JS를 올리면 되는데 파일명을 index.html로 해야 한다.
3. 풀 커밋 푸시하고 좀 기다렸다가 브라우저 주소창에 (프로젝트명).github.io 치고 접속하면 땡임.
유지보수가 어려운거 아니냐고요? 깃헙 저장소니까 그 안에 있는거 열고 수정하고 풀 커밋 푸시하고 좀 기다리면 반영됨.
'Coding > 코딩잡담' 카테고리의 다른 글
| 백준 섭종공지 (0) | 2026.04.15 |
|---|---|
| 안티그래비티를 써보았다. (0) | 2025.12.19 |
| CONNECT BY와 계층형 질의 (0) | 2025.11.14 |
| 스키마 (0) | 2025.11.14 |
| Select문에도 순서가 있다 (0) | 2025.11.13 |
어제 그 뼈대 올라왔잖아요? 그러고나서 밤에 잡아서 내용 다 채웠음. 농담같죠? 진짜임.
어바웃 페이지

뭘 많이 가렸죠? 다 개인정보라 어쩔 수 없음.. 그럼 논문하고 자격증은 왜 안 가렸냐… 논문은 이미 저널에 실린거고 자격증도 내가 땄다고 올렸잖아요. 쟤들은 이미 한번 깐거라 안 가린거임.
원래 생각했던 구성에는 저 카드가 없었습니다. 없었는데 제미나이가 "씁 근데 이거 이렇게만 보면 좀 심심하지 않을까요?라고 하면서 제안한겁니다. 저 선이랑 배경도 지금 조정중인데 선이 굵다고 이쁜건 아니고... 없애봤는데 이것도 아 씁 아닌 것 같고... 그림자는 그래도 배경이나 안이나 톤이 다 연해서 있는 게 나은 것 같긴 하고... 애매함. 그냥 애매함.
/* 개별 오브젝트를 감싸는 카드*/
.card {
width: 95%;
border: 1px solid var(--sub-dark);
border-radius: 10px;
margin: 10px 0;
padding: 10px;
background-color: var(--white);
box-shadow: 0 2px 6px rgba(0,0,0,0.08);
transition: all 0.2s ease;
}
.card:hover {
box-shadow: 0 6px 14px rgba(0,0,0,0.12);
transform: translateY(-2px);
}이렇게 트랜지션이랑 호버를 주면 마우스를 댔을 때 뜨는데 문제가 하나 있다. 여러분들 그거 아십니까? 모바일에서 호버 안되는거. 당연한 얘기지만, 서마터폰이나 태블릿 PC에서는 마우스를 쓰는 게 아니라 손으로 누르잖아요. 드래그도 손으로 하는데 어쨌든 이게 손으로 뭘 누른 상태에서 움직이고 걍 볼 때는 손을 또 안 대잖아요? 그래서 의미가 없다 이거요.
/*여러분 그 호버도 모바일에서는 끄셔야 하는 거 아십니까 */
@media (hover: hover) {
.card:hover {
box-shadow: 0 6px 14px rgba(0,0,0,0.12);
transform: translateY(-2px);
}
.accordion:hover {
background-color: var(--accent);
}
.link i:hover {
color: var(--accent);
transform: translateY(-1px) scale(1.05);
}
.btn:hover {
color: var(--accent);
}
}예. 미디어쿼리 주십시오.
근데 메인페이지는 이거 말고는 내용 말고 뭐 없는데 내용을 다 가려버려서 더 서술할게 없음.
스킬

어차피 헤더는 고정이니까 여기만 캡쳐하겠음... 이건 뭐냐면 내가 할 수 있는 것들을 나열한거다. 저 그리드 레이아웃은 나중에 스킬에 뭔가 더 추가되면 바뀔 수도 있는데 지금은 2열 2행이고 저기도 카드 적용되어있고 어이콘 폰트어썸이고... 그게 다임. 여기서 보고 가실건 저 체크입니다. 저게 li태그로 단 건데... 엥? li에 저런거 없는데요?
ul {
margin: 0;
list-style-position: inside;
list-style-type: none;
}
li::before {
content: "\f00c";
/* 7 버전 폰트 패밀리 확인 (보통 'Font Awesome 7 Free' 또는 'Font Awesome 7 Brands') */
font-family: "Font Awesome 7 Free";
font-weight: 900;
color: var(--sub-color);
margin: 0 10px;
}무늬를 빼고 ::before에 폰트어썸 달았음. 내가 포폴이라 체크표로 달긴 했는데 저거 응용하면 리스트에 이모지 다는 것도 가능합니다.
프로젝트

머여 삼색엔딩도 아니고 왜 색깔이 3개임? 그림을 잘 보시면 저기 초록색으로 된 부분은 아코디언 패널이 열려있죠? 그겁니다. 보라색이 기본이고 파란색은 당신이 마우스를 올리고 있는 패널, 초록색은 열어서 내용을 보고 있는 패널입니다. 근데 이렇게 놓고 보니 얘도 호버를 줄 필요는 없는 것 같아서 저 보고 있는 패널만 액센트로 빼야겠음.
저기는 아코디언 패널인데 지금까지 한 프로젝트들이 들어갑니다. 저기에는 생물쪽 한 거+부트캠프 팀플(아직 안 넣음)+생물학이 아닌 다른 EDA도 같이 들어가는데… 예, 그 캐글 EDA도 들어갑니다… 근데 여기서는 그림 안 넣고, 그냥 프로젝트에 대한 개요(이런거 했다)만 올린 다음 PDF를 올리거나 깃헙으로 유도할 예정임. 그래서 프로젝트 요얄이랑 과정만 있잖아요.
근데 왜 아코디언 패널로 함? 프로젝트 하나당 짧게짧게 한다 쳐도 이게 여러개거든요? 캐글도 선별해서 올리고 있지만 벌써 두개나 올라갔음… 이걸 그냥 보여주는건 좀 아닌 것 같음… 그래서 보고싶은 것만 펴서 보시라고 아코디언 패널로 넣은거다. 그럼 사진은요? 그래프 왜 뺌? 저게 이미지를 보여줄라면 서버에 그 이미지를 같이 올려야되거든요... 일단 그게 귀찮음...
참고로 깃헙이랑 태블로(폰트어썸에 아이콘이 없어서 저걸로 함)는 동작 잘 하는데 파일 링크는 동작을 안 합니다. 왜냐고? 링크를 파일로 안 걸었거든. 아직 파일이 안 올라간 상태입니다. 그리고 캐글 EDA나 캠블 EDA는 깃헙 링크만 있지 PDF도 없습니다. 개별 포폴을 안 만들었거든…
// 프로젝트 아코디언 패널
accordion.forEach((title) => {
title.addEventListener('click', () => {
// 클릭된 제목 바로 다음 요소(accordion_contents)를 타겟팅합니다.
const content = title.nextElementSibling;
const icon = title.querySelector('.icon-toggle');
title.classList.toggle('active');
// 보이고 안 보이고를 토글합니다.
if (content.style.display === 'block') {
content.style.display = 'none';
icon.style.transform = 'rotate(0deg)';
} else {
content.style.display = 'block';
icon.style.transform = 'rotate(180deg)';
}
});
});기존보다 업그레이드된 아코디언 패널 코드나 보고 가십셔.
1. 저 색깔이 울트라바이올렛이랑 비리디언으로 잡은건데 비리디언이 서브입니다. 서브 먼저 정하고 이색저색 해보다가 울트라바이올렛으로 한건데 얘가 채도가 그렇게 밝은 색이 아님. 좋게 말하자면 눈뽕이 없고 나쁘게 말하자면 그렇게 확 뛰는 색이 아닙니다. 그래서 그것때문에 고민을 많이 했음… 그런데 왜 보라색이죠? 내가 보라색 좋아함. 그 라미 다크라일락 몸통같은 보라색 좋아하는데… 팬톤아… 어떻게 2027년 색깔로 안되겠니…? 되겠냐
2. 액센트가 원래 골드컬러였는데 메인컬러가 보라색+서브컬러가 비리디언이라 액센트가 너무 붕 떠요. 그니까 그 색깔에 문제가 있는 게 아니라 그냥 안 맞는거임. 그래서 액센트를 파란색으로 한겁니다. 저게 비리디언+코발트블루 조합이었으면 아마 액센트로 골드컬러도 맞았을건데...
3. 헤더 오른쪽에 아이콘들 다 눌립니다. 진짜로 링크 걸어놨음.
4. 이걸 어디에 올릴지는 정해지지 않았는데, 이거 올려도 여기에 링크는 안 올릴듯합니다. 일단 저게 포폴이라 내 개인정보를 다 깠어요... 아까도 가리고 올렸잖음. 물론 이 글을 보고 있는 당신이 인사담당자고 내가 당신이 재직중인 회사에 지원하면서 이거 포폴인데 보실래요? 한다거나 나랑 링크드인 팔로워거나 하면 볼 수는 있겠지만 기본적으로 블로그에는 안 올릴 생각입니다. 지금 깃헙에는 올라가있는데 서버에서 내리고 나면 깃헙에서도 내릴까 생각중임.
5. CSS가 뭔가 많아지면서 주석의 소중함을 깨달았음… 주석이 없으니까 뭐가 뭔지 모르겠어요 이거… JS는 생각보다 뭐가 없는데 CSS가 어유 진짜 와… 이거 다 올리면 네이버가 짜름 5000자 넘는다고…
'Coding > JavaScript' 카테고리의 다른 글
| 달력에 기능 추가하기 (0) | 2026.04.21 |
|---|---|
| 제 2차 씨본 컬러 시뮬레이터 보수작업 (0) | 2026.04.16 |
| 포폴 웹페이지화-뼈대 잡기 (0) | 2026.03.07 |
| 개 얼탱이 없는 작업이 온다 두둥 (0) | 2026.03.06 |
| 씨본 팔레트 컬러 시뮬레이터 보수작업 (0) | 2026.02.22 |
내용이요? 이제 생각해야지.

여기까지 해서 뼈대가 잡혔고 탭 메뉴도 잘 돌아간다. 그럼 이제 내용물만 채우면 됩니다…
1. 저 언더 컨스트럭션 란에는 내 이름과 한줄 설명이 들어갈 예정이다. 그래서 일부러 넓게 안 잡았다. 꽉 차보여도 저 분량이 차지하는 건 가로 75%정도… (모바일은 미뎌쿼리 줘서 95%)
2. About에는 본인 약력, 자격증이 들어가고 skills는 뭐 할 수 있냐고 project에 개인적으로 했던 모든 프로젝트들이 들어갈 예정이다.

3. About 단을 하나로 할지 두개로 할 지 생각중임... 한쪽은 넓게 잡아서 내 소개 하고(아직 데이터쪽 약력이 없음...) 한쪽에 좀 좁게 잡아서 자격증이랑 외부 활동 내역같은 거 쓸까...
4. Project에 아코디언 패널 들어갑니다. 아코디언 패널이 뭐냐면 그 클릭하면 접히는? 그런거 있음. 프로젝트별로 패널 하나씩 할당됩니다.
5. HTML 코드는 어마무시하게 긴데 자바스크립트는 아직까진 그렇게 길지 않음… CSS……. (마른세수)
'Coding > JavaScript' 카테고리의 다른 글
| 제 2차 씨본 컬러 시뮬레이터 보수작업 (0) | 2026.04.16 |
|---|---|
| 포폴 웹페이지화-더 이상의 자세한 설명은 생략한다 (0) | 2026.03.08 |
| 개 얼탱이 없는 작업이 온다 두둥 (0) | 2026.03.06 |
| 씨본 팔레트 컬러 시뮬레이터 보수작업 (0) | 2026.02.22 |
| 씨본 컬러 파레트 씨뮬레이터 (0) | 2026.01.30 |

이게 뭐냐고요? 포폴을 웹으로 만들어서 서버에 올리자는 진심 얼탱이없는 작업에 들어갈 예정입니다. 근데 지금 팀플때문에 바빠서 구조 구상하고 색깔만 짜놨음.

대충 그렸음 대충...
1. 내 이름이랑 전번 이메일 깃헙 블로그(티스토리) 링크가 헤더에 들어가고(이부분도 고민 좀 해봐야됨...)
2. 그 밑에 내 이력이랑 스킬(뭐뭐 쓸 수 있나) 프로젝트가 들어가는데 이게 탭메뉴입니다. 그니까 얘를 탭하면 전환이 돼야 하는데 이걸 자바스크립트로 해야 하고…
3. 프로젝트에는 내 포폴에도 올라가는 프로젝트 세 개가 올라가는데 그거에 대한 설명을 개별 프로젝트로 아코디언 메뉴로 하고 PDF파일을 거기다가 올리든가 할겁니다.
링크 관련해서는 이걸 폰트어썸 아이콘만 넣을지(7.x로 올렸더만…) 이름을 병기할지정도 고민중임. 나머지는 뭐… 그렇죠 뭘.
'Coding > JavaScript' 카테고리의 다른 글
| 포폴 웹페이지화-더 이상의 자세한 설명은 생략한다 (0) | 2026.03.08 |
|---|---|
| 포폴 웹페이지화-뼈대 잡기 (0) | 2026.03.07 |
| 씨본 팔레트 컬러 시뮬레이터 보수작업 (0) | 2026.02.22 |
| 씨본 컬러 파레트 씨뮬레이터 (0) | 2026.01.30 |
| 특정 조건을 만족하면 DOM이 나타나게 해 보자 (0) | 2025.09.17 |