그렇다. 대망의 2부가 돌아왔다.
이게 정보 확인하는거 생략하고도 분량 꽤 되니까 알아서 쫓아오십쇼. 다음편에 태블로 얘기만 할거라서 이번편에 다 끝낼거임.
전처리
쓸 칼럼만 추리기
이게 칼럼이 되게 많은데 그걸 우리가 다 쓸 게 아니거든요? 그래서 쓸 것만 추린 다음에 데이터프레임을 재구성하고 그걸 csv파일로 보내야 합니다. 왜냐고? 그걸 보내야 태블로에서도 쓰죠.
analysis_column = ['CHROM','POS','REF','ALT','CLNSIG','CLNVC','GENEINFO','CLNREVSTAT'] # 칼럼 뭐하는건지 위에 있어요1. CHROM: 염색체(몇 번 염색체인지)
2. POS: 염색체 어디?
3. REF, ALT: 비포&애프터 (REF에 있는 시퀀스가 ALT로 바뀐 변이다)
4. CLNSIG: 임상적 유의성
5. CLNVC: 변이 타입(얘가 껴들어간겨 빠진겨 바뀐겨 뒤집어진겨)
6. GENEINFO: 유전자 이름+Entrez ID
7. 별점(왜 있는거냐)
이 칼럼들만 갖고와서
analysis_column = ['CHROM','POS','REF','ALT','CLNSIG','CLNVC','GENEINFO','CLNREVSTAT'] # 칼럼 뭐하는건지 위에 있어요
clinvar_df_analysis = clinvar_df[analysis_column]
clinvar_df_analysis이렇게 하면 데이터프레임 재구성은 끝난다.
그러고도 결측값이 왜 있는거냐고
이러고도 결측값이 있는 이유는 아직 연구가 덜 됐기 때문이다. 내가 isna()./sum()으로 확인해보고 원본까지 대조해본 결과임.
fill_values = {
'CLNSIG': 'Unknown_Significance',
'CLNREVSTAT': 'No_Assertion',
'GENEINFO': 'Unknown_Gene',
'MC': 'Unknown_Consequence'
}
clinvar_df_analysis = clinvar_df_analysis.fillna(value=fill_values)
clinvar_df_analysis['GENE_SYMBOL'] = clinvar_df_analysis['GENEINFO'].apply(
lambda x: x.split(':')[0] if ':' in x else x
)그래서 이게 최선이었습니다. 아, 하는 김에 유전자 이름도 분리함.
CLNSIG 범주화
# # 묶기 위한 조건 설정
conditions = [
clinvar_df_analysis['CLNSIG'].str.contains('Pathogenic|Likely_pathogenic', case=False, na=False),
clinvar_df_analysis['CLNSIG'].str.contains('Benign|Likely_benign', case=False, na=False),
clinvar_df_analysis['CLNSIG'].str.contains('Uncertain_significance|VUS', case=False, na=False),
clinvar_df_analysis['CLNSIG'].str.contains('Conflicting', case=False, na=False),
clinvar_df_analysis['CLNSIG'].str.contains('risk_factor|drug_response|association|protective|Affects', case=False, na=False)
]
# 결과 그룹명
choices = ['Pathogenic', 'Benign', 'VUS', 'Conflicting', 'Risk/Other']
# 기본값은 Unknown으로 설정
clinvar_df_analysis['CLNSIG_Group'] = np.select(conditions, choices, default='Unknown')
# 결과 확인
print(clinvar_df_analysis['CLNSIG_Group'].value_counts())나노 반도체 단위로 나뉘어진 CLNSIG을 대충 간소화했다. 여기까지 하고 to_csv로 저장해주면 태블로에서도 불러올 수 있습니다.
분석 드가자
CLNSIG별로 보기
clnsig_group = clinvar_df_analysis.groupby('CLNSIG_Group')['CLNSIG_Group'].count().sort_values(ascending=False)
clnsig_group
VUS는 말 그대로 몰?루인거고 Benign은 변이는 변이인데 누구나 하나쯤은 다 갖고 있는 뭐 그런거다. 그리고 그 다음으로 많은 게 Pathogenic이다.
ax = sns.barplot(clnsig_group)
plt.title('CLNSIG Group에 따른 변이 수')
plt.xlabel('CLNSIG Group')
plt.yscale('log') # 이거 안하면 막대기 하나 안보임
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, fmt='%d', padding=3, fontsize=10)
plt.tight_layout()
plt.show()
아… 나도 막대기 색을 나누고 싶었는데요… 아… 이게… size()로 했더니 시리즈가 돼서 애가 인식을 못해……
염색체 종류별 CLNSIG
아니 그럼 1번부터 다 보나요? 놉. 아까 내가 썼는지 모르겠는데 염색체도 범주화했다. 상염색체(1~22), 성염색체(XY), 미토콘드리아(얘는 지꺼 따로 있음)+언노운 이렇게 있음. 전에도 얘기했지만 1번부터 다 본다? 하나씩 그리면 스크롤이 너무 길고 그렇다고 모으자니 그래프가 뵈지도 않아요.
clnsig_chr_group = clinvar_df_analysis.groupby(['CHROM_Type','CLNSIG_Group']).size().unstack().fillna(0)
clnsig_chr_groupax = clnsig_chr_group.plot(kind='bar', stacked=True, ax=plt.gca(), color=sns.color_palette("Purples_r", n_colors=5))
plt.title('염색체 그룹에 따른 CLNSIG 그룹 수')
plt.xlabel('염색체')
plt.xticks(ticks=[0, 1, 2, 3], labels=['상염색체','미토콘드리아','성염색체','불명'])
plt.yscale('log') # 이거 안하면 막대기 하나 안보임
plt.tight_layout()
plt.show()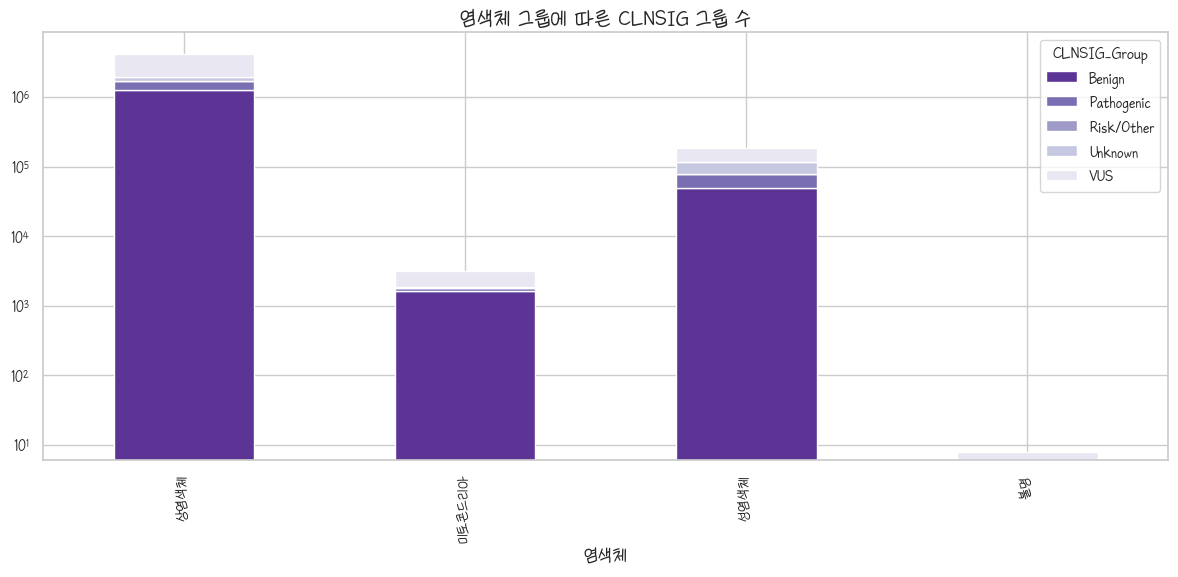
성염색체와 달리 상염색체와 미오콘드리아는 Benign 다음으로 VUS가 두드러지게 많다. 성염색체는 이렇게 보면 비슷비슷해보는데 차이가 존재하긴 함.
Pathogenic의 비중
clnsig_chr_group = clinvar_df_analysis.groupby(['CHROM_Type','CLNSIG_Group']).size().unstack().fillna(0)
clnsig_chr_group['total'] = clnsig_chr_group.sum(axis=1)
clnsig_chr_group['Pathogenic rate'] = round(clnsig_chr_group['Pathogenic'] / clnsig_chr_group['total'] * 100, 2)
clnsig_chr_group
뭔가… 비중이 생각보다 얼마 안됨… 아, Pathogenic은 발병시키는, 병원(호스피털 말고)성의 뭐 그런 뜻이다. 그니까 패소제닉한건 터지면 병되는건데 이게 유전자 바이 유전자지만 암이 되는 경우도 있고, 유전병이 되는 경우도 있다.
ax = sns.barplot(clnsig_chr_group, x = 'CHROM_Type', y = 'Pathogenic rate', hue='CHROM_Type')
plt.title('염색체별 Pathogenic 비율')
plt.xlabel('염색체')
plt.ylabel('Pathogenic 비율 (%)')
plt.xticks(ticks=[0, 1, 2], labels=['상염색체','미토콘드리아','성염색체'])
plt.xlim(-0.5, 2.5)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, fmt='%.2f', padding=3, fontsize=10)
# 얘는 로그 빼도 됩니다. 백분율이라;;
plt.tight_layout()
plt.show()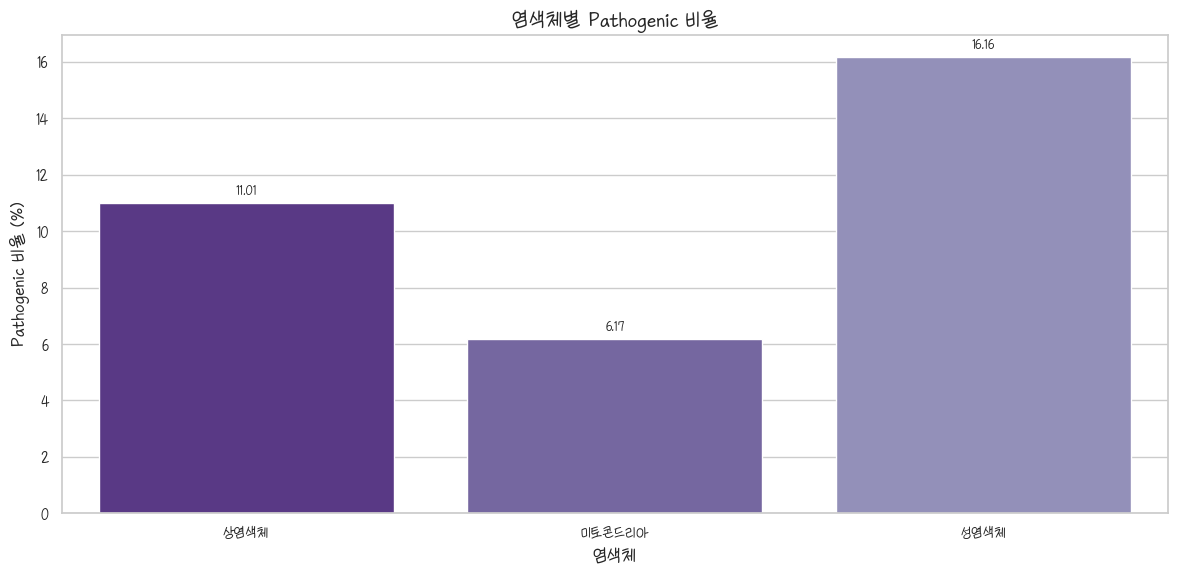
불명은 아예 0이라 축 조절하면서 빼버림… 저 축 틱이 0부터 2까지라고 범위도 0부터 2까지로 하시면 막대들이 양 옆으로 달라붙습니다. 항상 여유 범위를 주십시오.
Pathogenic한 변이들
clnsig_pathogenic = clinvar_df_analysis.query('CLNSIG == "Pathogenic"') # Pathogenic
clnsig_pathogenic이제 우리는 병원성 변이들에 주목해보자.
clnsig_pathogenic.groupby('CLNVC').size().sort_values(ascending=False)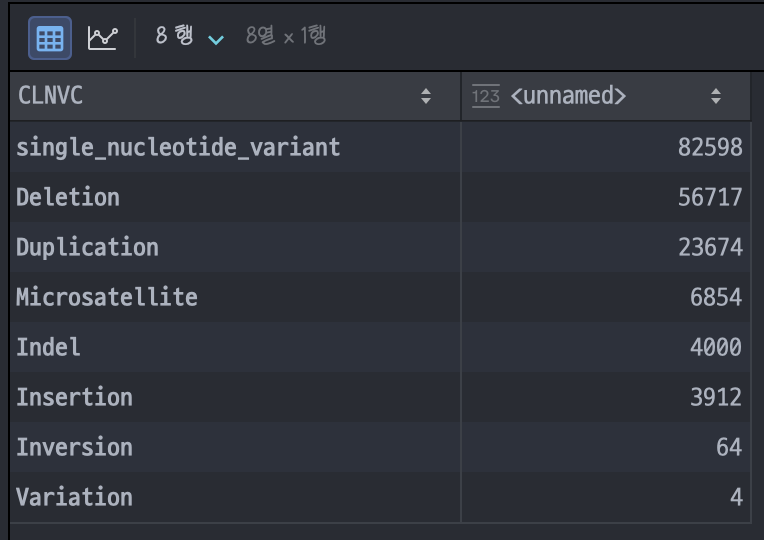
하필이면 제일 잡기 빡센 놈이 제일 많네… Single nucleotide variant가 뭐냐면… 그… 나비효과 아세요? 나비의 날갯짓이 지구 반대편에서 토네이도 된다는. 딱 그짝이다. 사람 몸 크기에 비하면 DNA 염기가 되게 작거든요. 염색체 안에 저런게 수백 수천만개가 때려박혀져 있는데 그 중에서 고거 하나 바뀐걸로 아미노산이 바뀌고(바꼈는데 같은 아미노산을 지정하는 경우도 있음) 그걸로 단백질 접힘이 바뀌고 접힘이 바뀐 단백질이 몸에 큰 영향을 끼친다.
저게 왜 잡기 빡세냐고? 스케일이 작잖아요. 염기가 뭉텅이로 빠진것도 아니고 딱 하나 빠진거잖아요. 마치 쌀알 10000개인 밥과 10001개인 밥을 주고 어떤게 만개게? 하는거랑 비슷하다.
ax = sns.barplot(clnsig_pathogenic_nvc)
purple_color = plt.colormaps['Purples'](0.2)
# 모든 막대(patch) 가져오기
for patch in ax.patches:
# 예: 특정 조건(height가 20 이상인 경우)의 막대만 색상 변경
if patch.get_height() > 20000:
pass
else:
patch.set_color(purple_color)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('변이 유형별 분포')
plt.ylabel('Count')
plt.show()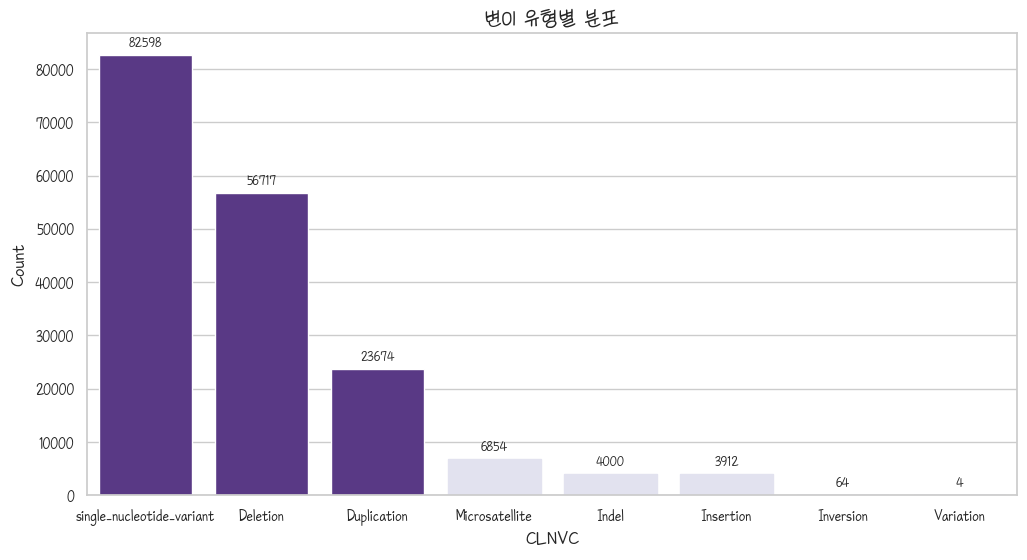
SNV는 알겠는데 저 뒤에 두개는 뭐냐… Deletion은 염기가 하나든 여러개든 있었는데 없었습니다 된 거고 Duplication은 시퀀스가 갑자기 원쁠원이 되는거다. 예를 들어서 GAATTC가 하나였다가 두개가 되는 거 말이다. transposon이랑은 다릅니다. 걔는 태생이 이사다니는 놈임.
염색체 유형별 분류
clnsig_pathogenic_chrom = clnsig_pathogenic.groupby('CHROM_Type').size().sort_values(ascending=False)
clnsig_pathogenic_chromax = sns.barplot(clnsig_pathogenic_chrom)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('염색체 종류별 분포')
plt.ylabel('Count')
plt.yscale('log')
plt.show()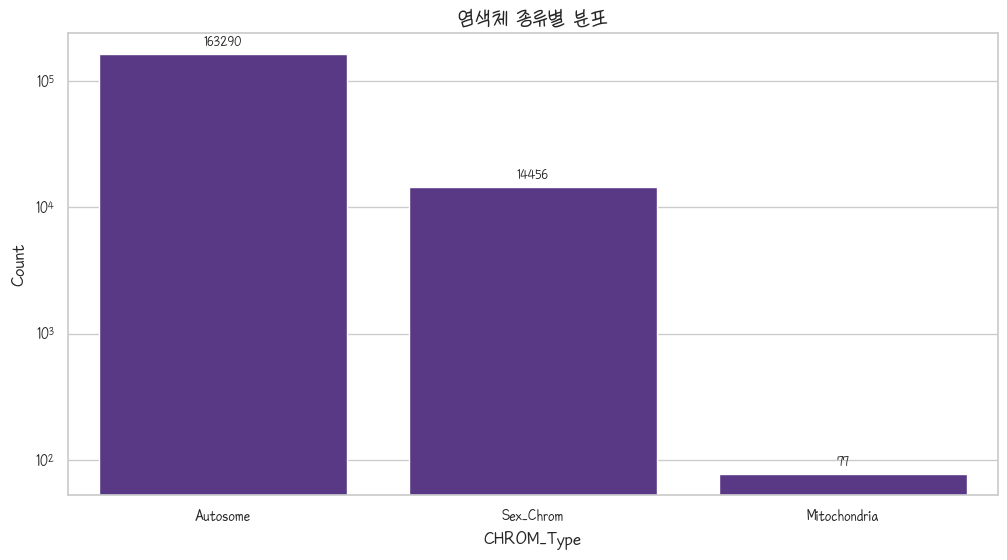
상염색체는 22개고(1~22) 성염색체는 두개라 그런가…?
염색체별 세분류
clnsig_pathogenic_chrom = clnsig_pathogenic.groupby('CHROM').size().sort_values(ascending=False)
clnsig_pathogenic_chromax = sns.barplot(clnsig_pathogenic_chrom[:10])
purple_color = plt.colormaps['Purples'](0.2)
# 모든 막대(patch) 가져오기
for patch in ax.patches:
# 예: 특정 조건(height가 20 이상인 경우)의 막대만 색상 변경
if patch.get_height() > 10000:
pass
else:
patch.set_color(purple_color)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('변이 유형별 분포 TOP 10 (염색체별)')
plt.ylabel('Count')
plt.show()
다 보기는 좀 거시기해서 TOP 10만 봤다. 17번, 2번 다음으로 X염색체가 많고 그 다음으로 1, 11번까지가 TOP 5다.
clnsig_pathogenic_chrom_Auto = clnsig_pathogenic.query('CHROM_Type == "Autosome"').groupby('CHROM').size().sort_values(ascending=False)
clnsig_pathogenic_chrom_Autoax = sns.barplot(clnsig_pathogenic_chrom_Auto[:10])
purple_color = plt.colormaps['Purples'](0.2)
# 모든 막대(patch) 가져오기
for patch in ax.patches:
# 예: 특정 조건(height가 20 이상인 경우)의 막대만 색상 변경
if patch.get_height() > 10000:
pass
else:
patch.set_color(purple_color)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('변이 유형별 분포 TOP 10 (상염색체)')
plt.ylabel('Count')
plt.show()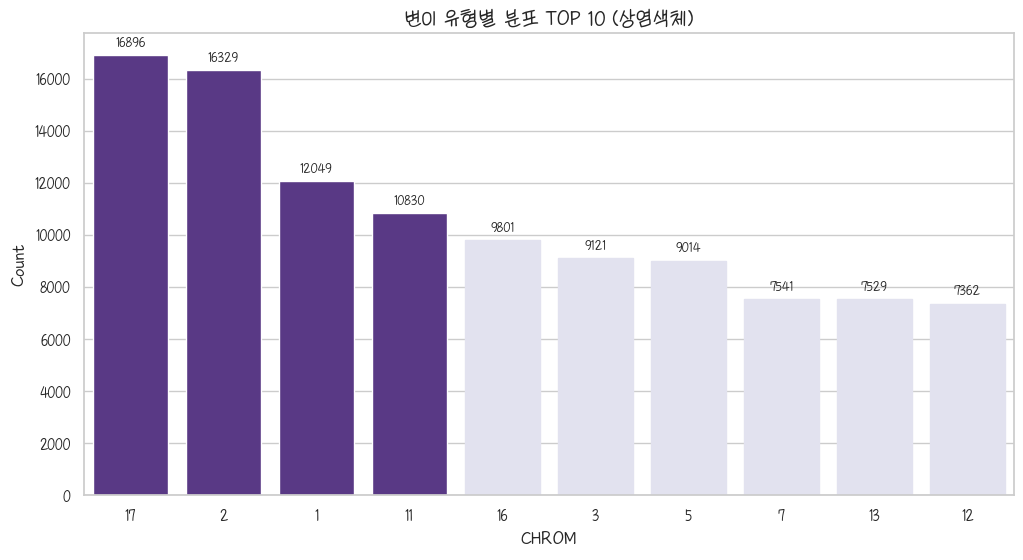
성염색체 빼고 상염색체만 봅시다. 17, 2, 1, 11, 16순으로 많다. 저거는 변이 개수가 10000개 이상인 것만 강조하는거라 저렇게 나온거임…
clnsig_pathogenic_chrom_Sex = clnsig_pathogenic.query('CHROM_Type == "Sex_Chrom"').groupby('CHROM').size().sort_values(ascending=False)
clnsig_pathogenic_chrom_Sexax = sns.barplot(clnsig_pathogenic_chrom_Sex)
purple_color = plt.colormaps['Purples'](0.2)
# 모든 막대(patch) 가져오기
for patch in ax.patches:
# 예: 특정 조건(height가 20 이상인 경우)의 막대만 색상 변경
if patch.get_height() > 10000:
pass
else:
patch.set_color(purple_color)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('변이 유형별 분포 TOP 10 (상염색체)')
plt.ylabel('Count')
plt.yscale('log')
plt.show()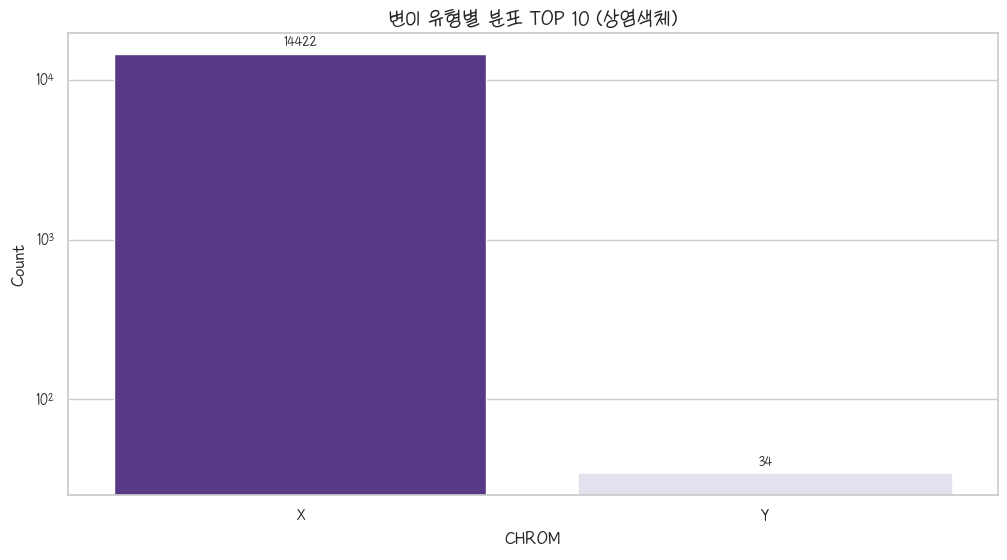
아놔 타이틀 수정해야되네.. 엥? X염색체가 더 많네요? 그 또한 체급차이다. Y염색체는 X염색체에 비해 짤똥하고 들어있는 유전자도 수십개정도지만, X염색체는 8~900개의 유전자가 들어있다.
유전자 TOP 10
clnsig_pathogenic_gene = clnsig_pathogenic.groupby('GENE_SYMBOL').size().sort_values(ascending=False)
clnsig_pathogenic_geneax = sns.barplot(clnsig_pathogenic_gene[:10])
# 모든 막대(patch) 가져오기
for patch in ax.patches:
# 예: 특정 조건(height가 20 이상인 경우)의 막대만 색상 변경
if patch.get_height() > 3500:
pass
else:
patch.set_color(purple_color)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('변이 유형별 분포 TOP 10 (유전자별)')
plt.xlabel('유전자')
plt.ylabel('Count')
plt.show()
BRCA 어디서 들어봤다 그죠? 안젤리나 졸리가 저 유전자에 변이가 있어서 유방을 절제하고 복원했잖음. 아니 왜 그렇게까지 해요? 저기 변이 있으면 유방암, 난소암에 걸릴 확률이 올라갑니다. 그러니까 암이라는 상태이상에 취약해지는 디버프인 셈이다. 이 변이도 유전되기때문에 아마 본인이 BRCA 변이가 있다면 가족중에 유방암이나 난소암에 걸린 사람이 계실 것이다.
그렇다고 어씨 나도 BRCA 변이 있네 조졌다 이럴것까진 없음. 우리는 늘 그렇듯이 여러분들이 최대한 건강하게 살 수 있도록 연구할겁니다.
SNV TOP 10
clnsig_pathogenic_snv = clnsig_pathogenic.query('CLNVC == "single_nucleotide_variant"').groupby('GENE_SYMBOL').size().sort_values(ascending=False)
clnsig_pathogenic_snvax = sns.barplot(clnsig_pathogenic_snv[:10])
# 모든 막대(patch) 가져오기
for patch in ax.patches:
# 예: 특정 조건(height가 20 이상인 경우)의 막대만 색상 변경
if patch.get_height() > 1000:
pass
else:
patch.set_color(purple_color)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('SNV가 가장 많은 유전자 TOP 10')
plt.xlabel('유전자')
plt.ylabel('Count')
plt.show()
NF1은 신경섬유종(1형)과 관련 있는 유전자고, FBN1은 찾아보니 피브릴린 1이란다. 마르판 증후군도 FBN1에 문제 생겼을 때 발생하는 병 중 하나다.
Deletion TOP 10
clnsig_pathogenic_del = clnsig_pathogenic.query('CLNVC == "Deletion"').groupby('GENE_SYMBOL').size().sort_values(ascending=False)
clnsig_pathogenic_delax = sns.barplot(clnsig_pathogenic_del[:10])
# 모든 막대(patch) 가져오기
for patch in ax.patches:
# 예: 특정 조건(height가 20 이상인 경우)의 막대만 색상 변경
if patch.get_height() > 1000:
pass
else:
patch.set_color(purple_color)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('Deletion이 가장 많은 유전자 TOP 10')
plt.xlabel('유전자')
plt.ylabel('Count')
plt.show()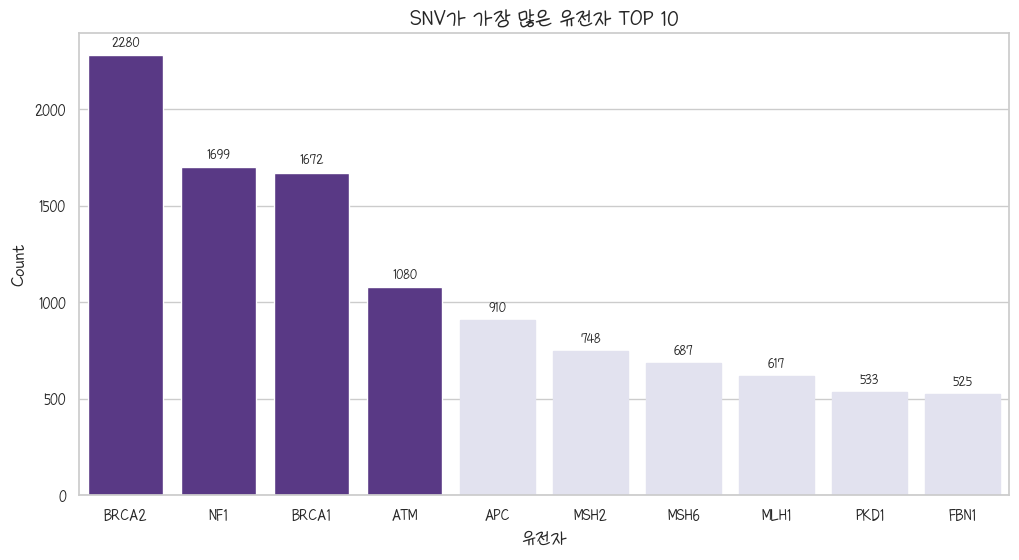
TOP 3은 어디서 많이 보셨던 애들이고… ATM은 찾아보니 암 억제 유전자다. 이건 또 뭐고? 암 억제 유전자는 발현되면 암을 막는 유전자고, 암 유전자(옹코진)는 발현되면 암 되는 유전자다. 전자는 브레이크, 후자는 엑셀.
Duplication TOP 10
clnsig_pathogenic_du = clnsig_pathogenic.query('CLNVC == "Duplication"').groupby('GENE_SYMBOL').size().sort_values(ascending=False)
clnsig_pathogenic_duax = sns.barplot(clnsig_pathogenic_du[:10])
# 모든 막대(patch) 가져오기
for patch in ax.patches:
# 예: 특정 조건(height가 20 이상인 경우)의 막대만 색상 변경
if patch.get_height() > 500:
pass
else:
patch.set_color(purple_color)
for container in ax.containers:
# fmt='%d'는 정수로 표시, label_type='edge'는 막대 끝에 표시
ax.bar_label(container, padding=3, fontsize=10)
plt.title('Duplication이 가장 많은 유전자 TOP 10')
plt.xlabel('유전자')
plt.ylabel('Count')
plt.show()
일단 여기까지 하고 태블로로 빠졌음.
'Coding > EDA' 카테고리의 다른 글
| Google Play Store – Most Downloaded Android Apps (0) | 2026.03.02 |
|---|---|
| 얘! clinvar도 EDA가 된단다! (3) (0) | 2026.02.18 |
| 얘! clinvar도 EDA가 된단다! (1) (0) | 2026.02.15 |
| Ramen ratings (0) | 2026.02.11 |
| Post-COVID Video Games Worldwide (2021-2025) (0) | 2026.02.10 |