나도 이걸 4번까지 하게 될 줄은 몰랐음…
개요
여러분들 다들 아시죠? 코로나19가 우리를 어떻게 변화시켰는지...
저는 자가격리도 해보고 걸려도 봤습니다. 자가격리때 다들 헐 어카냐 했는데… 저는 태생이 집순이라 1도 데미지 없었고요.. 격리소가 을지로 근처라 원격으로 명동성당 루기아 줘팼습니다.
창고는 언제나 열려있소
# 쿼리 조건: SARS-CoV-2 (코로나19), Spike 단백질 위주로 털어보기
# 2025년 최신 데이터 + 사람 숙주 조건
query = "SARS-CoV-2 AND S[Gene Name] AND 2025[PDAT] AND Homo sapiens[Host]"
# 1. ID 리스트 가져오기
handle = Entrez.esearch(db="nucleotide", term=query, retmax=300)
record = Entrez.read(handle)
id_list = record["IdList"]
handle.close()
# 2. 실제 서열 데이터 가져오기 (FASTA 형식)
fetch_handle = Entrez.efetch(db="nucleotide", id=id_list, rettype="fasta", retmode="text")
sequences = list(SeqIO.parse(fetch_handle, "fasta"))
fetch_handle.close()
# 3. 저장
with open("influenza_h3n2.fasta", "w") as f:
SeqIO.write(sequences, f, "fasta")
print(f"성공적으로 {len(sequences)}개의 서열을 가져왔습니다.")
print("----------")
for record in sequences[:3]:
print(f"ID: {record.id}")
print(f"Description: {record.description}")
print(f"Length: {len(record.seq)} bp\n")...근데 저걸 꼭 2025년만 가져오는 이유가 있는거니 제미나이야...?
# 콤퓨타에 저-장
vir_sequence = []
for i, id in enumerate(id_list):
print(f"Downloading sequence {i+1}/{len(id_list)}: {id}")
handle = Entrez.efetch(db="nucleotide", id=id, rettype="fasta", retmode="text")
record = SeqIO.read(handle, "fasta")
# record에 id와 seq가 다 들어가야되더라... (안되면 오류남 봤음)
vir_sequence.append(record)
# 파일로 저장
SeqIO.write(vir_sequence, "hantavirus_sequence.fasta", "fasta")
print('Done!')# 시퀀스 길이 체크
for rec in vir_sequence:
print(f"ID: {rec.id} | Length: {len(rec.seq)}")이게 그… 체크해봤더니 완전 잡탕찌개인거예요… 뭔 닥터 스트레인지 왔다감도 아니고 대혼돈의 유니버스여 게놈이… 저게 전체 데이터는 한 다섯자리 되고, 중간중간 3~4000bp정도 되는게 바이러스 스파이크(그 겉면에 삐죽삐죽한 작대기 있음)인데, 저걸 저대로 MSA하면 일단 똥때리고 밥먹고 자다 인나서 다음날이 됐는데도 MSA가 안 끝났을 확률이 99.9%입니다. 그리고 우리 스파이크만 할거임.
# 가져온 sequences 리스트에서 길이로 필터링
spike_only = [rec for rec in sequences if 3000 <= len(rec.seq) <= 4500]
# 필터링된 결과 저장
with open("sars_cov2_spike_clean.fasta", "w") as f:
SeqIO.write(spike_only, f, "fasta")
print(f"전체 {len(sequences)}개 중 진짜 스파이크만 {len(spike_only)}개 골라냈습니다.")그래서 어떻게 해요? 아 스파이크만 거르셔야죠.
MSA
print('MSA start... ')
# MSA 분석 시-작
try:
result = subprocess.run([muscle_exe, "-align", "sars_cov2_spike_clean.fasta", "-output", "sars_cov2_spike_align.fasta"], check=True, capture_output=True, text=True)
print("Completed. ")
except subprocess.CalledProcessError as e:
print(f"MSA failed: {e}")
finally:
alignment = AlignIO.read("influenza_h3n2_muscle_aligned.fasta", "fasta")
# 밥 먹고 오면 끝나있겠는데...?보통 돌려놓고 똥때리고 오면 끝나있습니다. 담배는 안되냐고요? 뭐 알아서 피시든지... 나한테 냄새만 안 오게 하십쇼.
print("====== MSA Result ======")
alignment = AlignIO.read("sars_cov2_spike_align.fasta", "fasta") # FASTA 니네 확장자가 몇개냐...
for record in alignment:
print(f"{record.id[:10]:<15} : {record.seq[:100]}")====== MSA Result ======
PV687028.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687026.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687025.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687024.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687027.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAAATCAATC---------ATACACTA
PV687011.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687009.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687016.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687015.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687014.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687012.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687023.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687022.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687019.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687010.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687013.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687021.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687020.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687018.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PV687017.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTCATGCCGCTGTTTAATCTTATAACTACAACTCAATC---------ATACACTA
PX775888.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGT------------TAATCTTAGAACCAGAACTCAATTACCCCCTGCATACACTA
PX765932.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGT------------TAATCTTACAACCAGAACTCAATTACCCCCTGTATATATTA
PV687008.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGT------------TAATCTTATAACCAGAACTCAATC---------ATACACTA
PV687007.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGT------------TAATCTTATAACCAGAACTCAATC---------ATACACTA
PX765940.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGT------------TAATCTTACAACCAGAACTCAATTACCCCCTGCATACACTA
PX736381.1 : ATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGT------------TAATCTTACAACCAGAACTCAATTACCCCCTGTATATATTA다시 말씀드리는거지만 저 작대기는 갭이고요... 여 염기 읍따! 입니다... 예...
평균 보존율
def calculate_conservation_no_gap(alignment, gap_threshold=0.5):
length = alignment.get_alignment_length()
scores = []
for i in range(length):
column_raw = alignment[:, i]
# gap 비율이 너무 높으면 제외 (선택사항)
gap_fraction = column_raw.count("-") / len(column_raw)
if gap_fraction > gap_threshold:
continue
# gap 제거
column = column_raw.replace("-", "")
if len(column) == 0:
continue
# 최빈 염기 비율 = 보존도
most_common = max(set(column), key=column.count)
score = column.count(most_common) / len(column)
scores.append(score)
return scores
scores = calculate_conservation_no_gap(alignment)
print(f"해당 구간의 평균 보존율: {np.mean(scores)*100:.2f}%")해당 구간의 평균 보존율: 99.53%너 바이러스잖아...
섀넌 엔트로피
plt.figure(figsize=(15, 7))
plt.plot(variation_scores, alpha=0.8, color='#00498c')
plt.fill_between(range(len(variation_scores)), variation_scores, alpha=0.3)
plt.axvline(1322, linestyle='--', color='red', alpha=0.5)
plt.text(1322, max(variation_scores)*1.07, 'Spike POS 1322', color='red', alpha=0.5, ha='center', fontweight='bold')
plt.axvline(3295, linestyle='--', color='red', alpha=0.5)
plt.text(3295, max(variation_scores)*1.07, 'Spike POS 3295', color='red', alpha=0.5, ha='center', fontweight='bold')
plt.axvline(626, linestyle='--', color='red', alpha=0.5)
plt.text(626, max(variation_scores)*1.07, 'Spike POS 626', color='red', alpha=0.5, ha='center', fontweight='bold')
plt.title("Viral Variation Hotspots", y = 1.07)
plt.xlabel("Alignment Position (filtered)")
plt.ylabel("Normalized Shannon Entropy")
plt.show()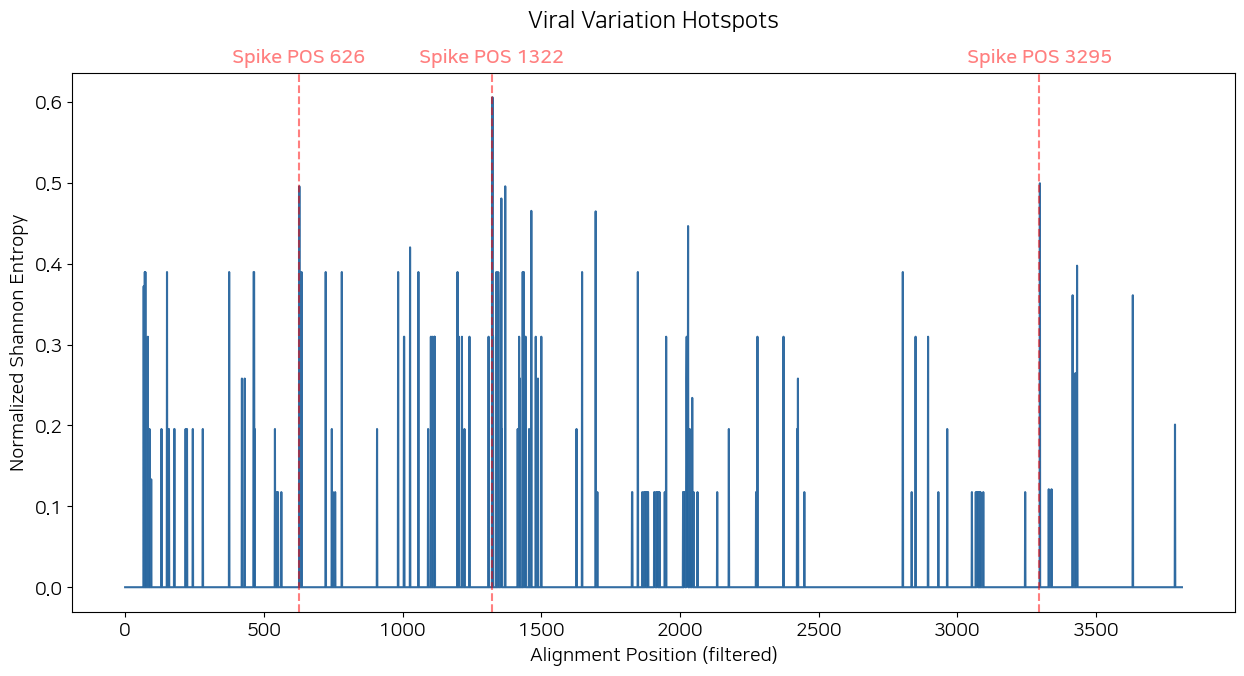
저 그래프 코드에 axvline 세 개 달려있는게 핫스팟 TOP 3임. 저걸 한타 인플루엔자 라이노바이러스까지 다 넣느라 좀 걸렸습니다... 솔직히 있으면 좋잖아.
# 섀넌 엔트로피 점수 도출
def get_top_variable_sites_no_gap(alignment, top_n=10):
length = alignment.get_alignment_length()
variability = []
ref_seq = alignment[0].seq
for i in range(length):
# 🔴 reference가 gap이면 무조건 스킵
if ref_seq[i] == '-':
continue
column = alignment[:, i].replace("-", "")
if not column:
continue
counts = Counter(column)
total = sum(counts.values())
entropy = 0.0
for c in counts.values():
p = c / total
entropy -= p * math.log2(p)
variability.append((i, entropy))
return sorted(variability, key=lambda x: x[1], reverse=True)[:top_n]
def alignment_to_sequence_pos(aligned_seq, aln_pos):
count = 0
for i in range(aln_pos + 1):
if aligned_seq[i] != '-':
count += 1
return count
ref_seq = alignment[0].seq
top_sites = get_top_variable_sites_no_gap(alignment, top_n=10)
high_entropy_ha_sites = []
print("--- 변이가 집중된 주요 포지션 분석 결과 ---")
for aln_pos, score in top_sites:
real_pos = alignment_to_sequence_pos(ref_seq, aln_pos)
high_entropy_ha_sites.append(real_pos)
print(f"Alignment {aln_pos:4d} → Spike Pos {real_pos:4d} | 엔트로피: {score:.3f}")
print("\n최종 고엔트로피 포지션 리스트:")
print(high_entropy_ha_sites)--- 변이가 집중된 주요 포지션 분석 결과 ---
Alignment 1345 → Spike Pos 1322 | 엔트로피: 1.212
Alignment 3321 → Spike Pos 3295 | 엔트로피: 0.999
Alignment 649 → Spike Pos 626 | 엔트로피: 0.991
Alignment 1391 → Spike Pos 1368 | 엔트로피: 0.991
Alignment 1377 → Spike Pos 1354 | 엔트로피: 0.961
Alignment 1488 → Spike Pos 1462 | 엔트로피: 0.931
Alignment 1720 → Spike Pos 1694 | 엔트로피: 0.929
Alignment 2053 → Spike Pos 2027 | 엔트로피: 0.893
Alignment 1048 → Spike Pos 1025 | 엔트로피: 0.840
Alignment 3455 → Spike Pos 3429 | 엔트로피: 0.795
최종 고엔트로피 포지션 리스트:
[1322, 3295, 626, 1368, 1354, 1462, 1694, 2027, 1025, 3429]저기서 엔트로피가 가장 높은 포인트 세 개를 axvline으로 넣은겁니다. 가릿?
맨-휘트니 U 검정
# --- 2. normalization ---
entropy_min = np.nanmin(entropy_window)
entropy_max = np.nanmax(entropy_window)
entropy_window_normalized = (
entropy_window - entropy_min
) / (entropy_max - entropy_min)
variation_scores = entropy_window_normalized
# --- 3. NaN 제거 (🔥 중요) ---
valid_scores = variation_scores[~np.isnan(variation_scores)]
# --- 4. hotspot threshold (normalized 기준) ---
threshold_norm = np.percentile(valid_scores, 90)
threshold_raw = threshold_norm * (entropy_max - entropy_min) + entropy_min
hotspots = valid_scores[valid_scores >= threshold_norm]
non_hotspots = valid_scores[valid_scores < threshold_norm]
u_stat, p_value = mannwhitneyu(
hotspots,
non_hotspots,
alternative="greater"
)
print(f"Hotspot threshold (top 10%, normalized entropy): {threshold_norm:.3f}")
print(f"Corresponding raw entropy threshold: {threshold_raw:.3f}")
print(f"Mann–Whitney U statistic: {u_stat:.1f}")
print(f"p-value: {p_value:.4e}" if p_value > 1e-10 else "p-value: <1e-10")Hotspot threshold (top 10%, normalized entropy): 0.235
Corresponding raw entropy threshold: 0.056
Mann–Whitney U statistic: 1309496.0
p-value: <1e-10거 깃헙 가보시면 통계값도 있으니까 함 보세요... 아무튼 귀무가설(코로나바이러스 스파이크의 변이는 무작위적으로 발생하며, 특정 위치에 선호적으로 집중되지 않는다.) 압도적 기각. 근데 시각화한 거 보셨으면 대충 귀무가설 기각될 것 같긴 했죠? 그래프에 수평선이 보이니까.
Phylogenic tree
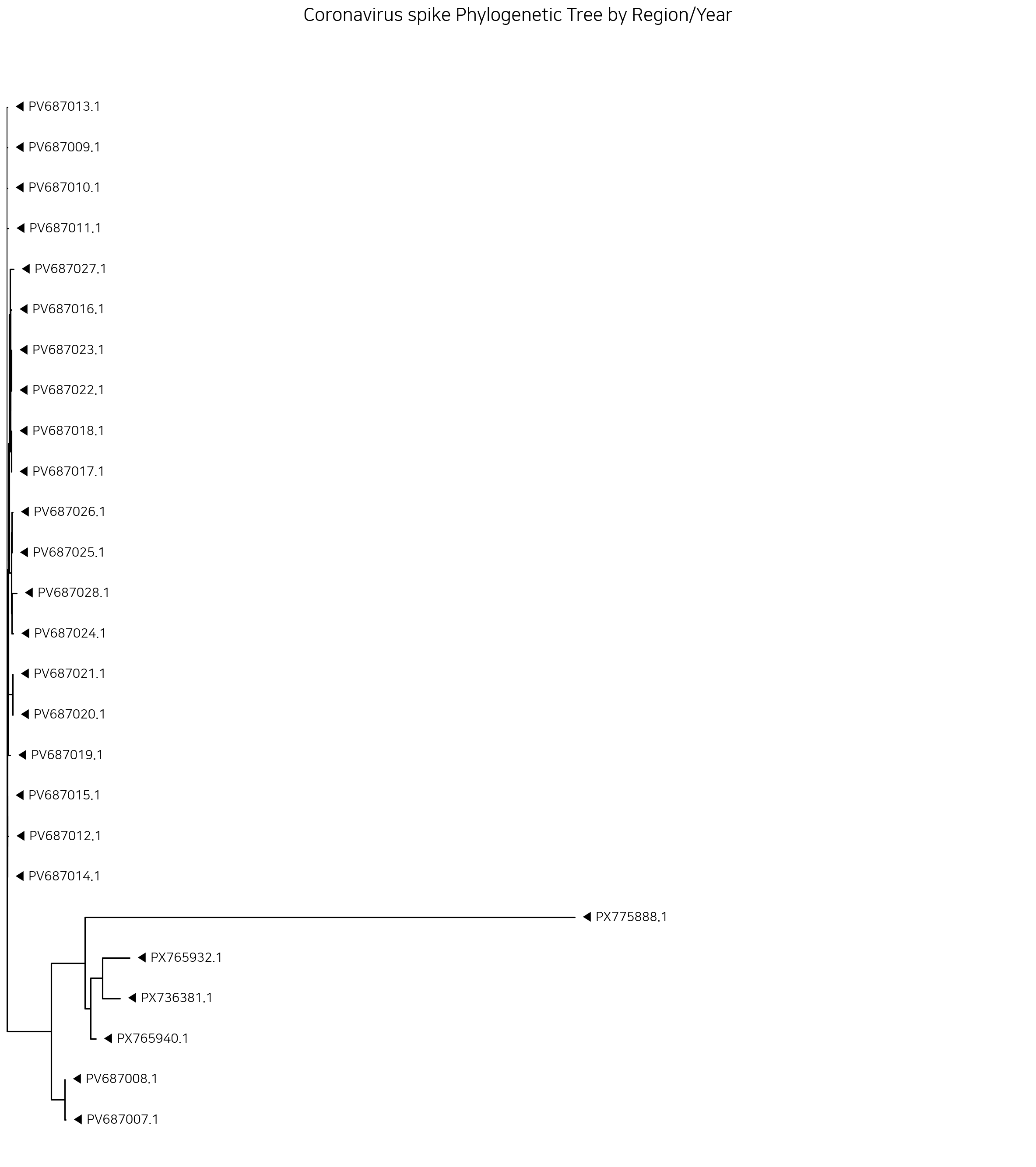
코드는 네이버에서 짤릴까봐 뺐습니다. 이것도 깃헙가서 보세요. 근데 트리 코드는 딱히 바뀐게 없음…
스피어맨 상관계수
그 우리 인플루엔자 할 때도 트리 관련해서 맨 휘트니 안했죠? 그때 내가 걔들은 같은 아종에서 바뀌는거라 맨 휘트니 하면 결과 이상하게 나온다고 했잖아요. 근데 얘네도 비슷해요 이유는. 스파이크 단백질이 어어하면 숙주 찾아서 입장하기도 전에 뽀사지거든… 그래서 보존구역이 많은거고, 그래서 맨 휘트니를 못합니다.
rho, p = spearmanr(tree_distances, seq_identities)
print(f'Rho: {rho:.4f}')
print(f'P-value: {p:.4e}')Rho: -0.9950
P-value: 0.0000e+00얘도 음의 상관관계네 ㅋㅋㅋㅋ
plt.figure(figsize=(10, 7), dpi=120)
sns.regplot(x=tree_distances, y=seq_identities,
scatter_kws={'alpha':0.2, 'color':'gray', 's':10},
line_kws={'color':'red', 'label': f'Spearman Rho: {rho:.3f}'})
plt.title("Coronavirus: Tree Distance vs Sequence Identity", fontsize=15, pad=15)
plt.xlabel("Genetic Distance on Tree", fontsize=12)
plt.ylabel("Pairwise Sequence Identity", fontsize=12)
plt.legend()
plt.grid(True, linestyle='--', alpha=0.5)
plt.show()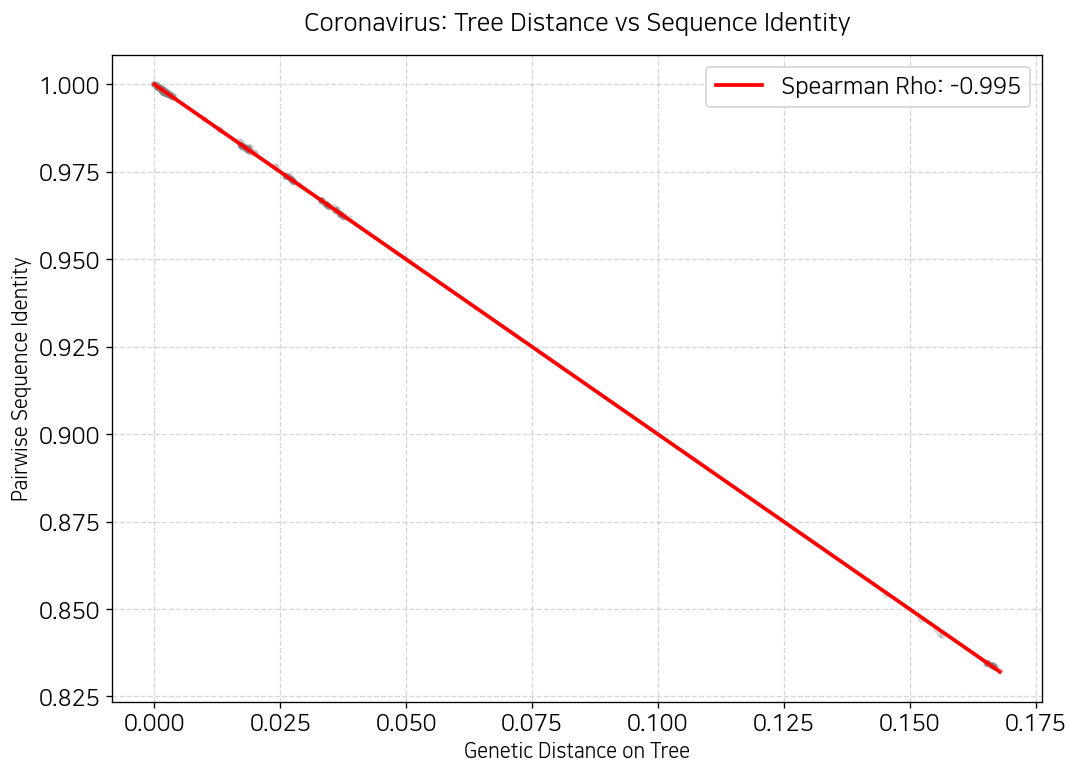
오… 이게 봐봐요. 계통수에서 거리가 멀수록 진화적으로 먼? 뭐 그런거잖아요? 그래서 스피어만 상관계수가 음의 상관계수가 나온거다. 트리의 거리가 멀어질수록 유사성도 떨어지잖아요.
'Coding > Python' 카테고리의 다른 글
| MSA에 군집분석을 끼얹어보세요! (0) | 2026.02.20 |
|---|---|
| M1V1 = M2V2 (0) | 2026.02.15 |
| 라이노바이러스 유전자로 MSA를 해보았다 (0) | 2026.01.27 |
| 식물 데이터도 분석이 되나요? (0) | 2026.01.27 |
| 매우 주관적인 씨본 컬러맵 고르는 방법 (0) | 2026.01.26 |