쿡북 분량 개짧음 진짜 이거보다 짧을수가 없음.
KEGG?
KEGG: Kyoto Encyclopedia of Genes and Genomes
www.genome.jp
Kyoto Encyclopedia of Genes and Genomes의 준말. 그렇다, 이름에 교토가 들어간 걸 보면 아시겠지만 일제 DB다.

여기가 메인페이지

여기가 KEGG brite. 본인이 자주 가는 곳이다. 생각보다 쏠쏠한 정보가 많고 KEGG brite의 경우 golden standard dataset이라고 해서 야먀니시가 인공지능 학습시킬 때 쓴 데이터셋(GPCR, 효소, 핵 리셉터, 트랜스포터) 분류별로 약물 타겟을 알려준다.
Parsing
파싱할거면 일단 파일이 당신 컴퓨터에 있어야 한다.
급한대로 이거라도 쓰자. (아님 밑에 쿼링에서 갖고오든가...)
from Bio.KEGG import Enzyme
records = Enzyme.parse(open("/home/koreanraichu/ec_5.4.2.2.txt"))
record = list(records)[0]
print(record)전체를 뽑고 싶으면 이렇게 하고
from Bio.KEGG import Enzyme
records = Enzyme.parse(open("/home/koreanraichu/ec_5.4.2.2.txt"))
record = list(records)[0]
print(record.classname)['Isomerases;', 'Intramolecular transferases;', 'Phosphotransferases (phosphomutases)']특정 부분만 보고 싶으면 이렇게 한다. 근데 쟤 뭐 하는 효소냐...
아, parse 친구 read도 있다. 데이터가 단식이면 리드, 뭉텅이면 파스.
Querying
쿼리에 ing면 쿼링인가...
Enzyme
from Bio.KEGG import REST
from Bio.KEGG import Enzyme
request = REST.kegg_get("ec:7.1.2.2")
# 참고: ATP synthase의 EC번호이다
open("ec_7.1.2.2.txt", "w").write(request.read())
records = Enzyme.parse(open("ec_7.1.2.2.txt"))
record = list(records)[0]
print(record)참고: EC 7.1.2.2는 ATP synthase다

파일이 생긴 것을 볼 수 있다. (본인은 Pycharm으로 해서 파이참 경로에 생겼다)
Pathway
사실상 KEGG의 근본은 pathway data다. 정말 세상 온갖가지 pathway가 다 있음. 진짜로.
from Bio.KEGG import REST
human_pathways = REST.kegg_list("pathway", "hsa").read()
print(human_pathways)뭔지 모르겠을 땐 일단 다 불러오는 게 국룰이다. 밑에 보여주겠지만 pathway만 써도 불러오는 건 된다.
from Bio.KEGG import REST
human_pathways = REST.kegg_list("pathway", "hsa").read()
# 여기까지 분량이 정말 많은 것을 볼 수 있다.
repair_pathways = []
for line in human_pathways.rstrip().split("\n"):
entry, description = line.split("\t")
if "repair" in description:
repair_pathways.append(entry)
print(repair_pathways)
# 조건부로 Repair pathway만 보는 코드['path:hsa03410', 'path:hsa03420', 'path:hsa03430']다 좋은데 그걸 꼭 hsa로 뽑아야겠냐... 알아서 찾아라 이거지 이럴거면 KEGG가지 뭐하러 여기서 찾냐
from Bio.KEGG import REST
human_pathways = REST.kegg_list("pathway", "hsa").read()
# 여기까지 분량이 정말 많은 것을 볼 수 있다.
repair_pathways = []
for line in human_pathways.rstrip().split("\n"):
entry, description = line.split("\t")
if "repair" in description:
repair_pathways.append(description)
print(repair_pathways)
# 조건부로 Repair pathway만 보는 코드['Base excision repair - Homo sapiens (human)', 'Nucleotide excision repair - Homo sapiens (human)', 'Mismatch repair - Homo sapiens (human)']아니 그럼 출력을 description으로 바꾸면 되잖아. 와 천잰데?
from Bio.KEGG import REST
human_pathways = REST.kegg_list("pathway", "hsa").read()
# 여기까지 분량이 정말 많은 것을 볼 수 있다.
repair_pathways = []
for line in human_pathways.rstrip().split("\n"):
entry, description = line.split("\t")
if "*sis" in description:
repair_pathways.append(description)
print(repair_pathways)
# 조건부로 Repair pathway만 보는 코드[]와일드카드는 안먹거나 *이 아닌 다른 기호인 듯 하다.
Gene
Pathway에서 이어진다.
from Bio.KEGG import REST
human_pathways = REST.kegg_list("pathway", "hsa").read()
# 여기까지 분량이 정말 많은 것을 볼 수 있다.
repair_pathways = []
for line in human_pathways.rstrip().split("\n"):
entry, description = line.split("\t")
if "repair" in description:
repair_pathways.append(entry)
# 조건부로 Repair pathway만 보는 코드
# 여기까지는 파일에 있었으니까 다들 아시리라 믿고...
repair_genes = []
for pathway in repair_pathways:
pathway_file = REST.kegg_get(pathway).read() # query and read each pathway
# iterate through each KEGG pathway file, keeping track of which section
# of the file we're in, only read the gene in each pathway
current_section = None
for line in pathway_file.rstrip().split("\n"):
section = line[:12].strip() # section names are within 12 columns
if not section == "":
current_section = section
if current_section == "GENE":
gene_identifiers, gene_description = line[12:].split("; ")
gene_id, gene_symbol = gene_identifiers.split()
if not gene_symbol in repair_genes:
repair_genes.append(gene_symbol)
print(
"There are %d repair pathways and %d repair genes. The genes are:"
% (len(repair_pathways), len(repair_genes))
)
print(", ".join(repair_genes))일단 코드가 긴데
1. Pathway 찾아주는 코드(그래서 entry로 쳐야 먹힌다)
2. 에서 Gene 섹션 찾아서 출력해준다. (hsa 번호가 맞는 pathway에서 gene을 찾아준다)
There are 3 repair pathways and 78 repair genes. The genes are:
OGG1, NTHL1, NEIL1, NEIL2, NEIL3, UNG, SMUG1, MUTYH, MPG, MBD4, TDG, APEX1, APEX2, POLB, POLL, HMGB1, XRCC1, PCNA, POLD1, POLD2, POLD3, POLD4, POLE, POLE2, POLE3, POLE4, LIG1, LIG3, PARP1, PARP2, PARP3, PARP4, FEN1, RBX1, CUL4B, CUL4A, DDB1, DDB2, XPC, RAD23B, RAD23A, CETN2, ERCC8, ERCC6, CDK7, MNAT1, CCNH, ERCC3, ERCC2, GTF2H5, GTF2H1, GTF2H2, GTF2H2C_2, GTF2H2C, GTF2H3, GTF2H4, ERCC5, BIVM-ERCC5, XPA, RPA1, RPA2, RPA3, RPA4, ERCC4, ERCC1, RFC1, RFC4, RFC2, RFC5, RFC3, SSBP1, PMS2, MLH1, MSH6, MSH2, MSH3, MLH3, EXO1그래서 이렇게 나온다.
근데 솔직히 인간적으로 저것만 찾으면 개노잼 아니냐. 그래서 준비했다.
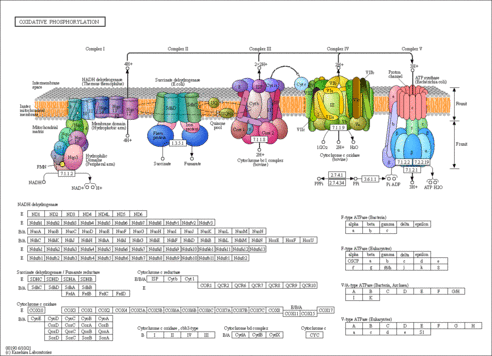
이건 산화적 인산화(Oxidative phosphorylation)인데, ATP를 얻기 위한 과정 중 하나이다. 여러분들이 일반적으로 알고 있는 ATP 생산 절차는
1. 우리가 먹은 포도당을 해당과정(Glycolysis)을 통해 피루브산 두 개를 만들고
2. TCA cycle(크렙 회로나 시트르산 사이클이라고도 한다) 들어가고(여기서부터 미토콘드리아 영역)
3. Oxidative phosphorylation을 통해 미토콘드리아가 ATP를 만든다(여기서 겁나 나온다)
이건데... Oxidative phosphorylation은 마지막 절차이다. 보통 TCA cycle부터 미토콘드리아 영역이고 피루브산이 TCA cycle로 가면 돌아올 수 없는 강을 건너게 된다. (피루브산 상태에서 포도당신생합성(Gluconeogenesis)을 돌리기도 한다. 번역명 왜이래요. 번역 간지나네)
여담이지만 과당도 해당과정 중간에 낄 수 있다. (Galactose도 낀다...젖당이었나 갈락이었나 아무튼 낌)
from Bio.KEGG import REST
human_pathways = REST.kegg_list("pathway", "hsa").read()
# 여기까지 분량이 정말 많은 것을 볼 수 있다.
Oxidative_pathways = []
Oxidative_pathways_name =[]
for line in human_pathways.rstrip().split("\n"):
entry, description = line.split("\t")
if "Oxidative" in description:
Oxidative_pathways.append(entry)
Oxidative_pathways_name.append(description)
print("Entry no: ",Oxidative_pathways,"description is: ",Oxidative_pathways_name)
# 조건부로 Repair pathway만 보는 코드
# 여기까지는 파일에 있었으니까 다들 아시리라 믿고...
Oxidative_genes = []
for pathway in Oxidative_pathways:
pathway_file = REST.kegg_get(pathway).read() # query and read each pathway
# iterate through each KEGG pathway file, keeping track of which section
# of the file we're in, only read the gene in each pathway
current_section = None
for line in pathway_file.rstrip().split("\n"):
section = line[:12].strip() # section names are within 12 columns
if not section == "":
current_section = section
if current_section == "GENE":
gene_identifiers, gene_description = line[12:].split("; ")
gene_id, gene_symbol = gene_identifiers.split()
if not gene_symbol in Oxidative_genes:
Oxidative_genes.append(gene_symbol)
print(
"There are %d pathways and %d genes. The genes are:"
% (len(Oxidative_pathways), len(Oxidative_genes))
)
print(", ".join(Oxidative_genes))Entry no: ['path:hsa00190'] description is: ['Oxidative phosphorylation - Homo sapiens (human)']
There are 1 pathways and 134 genes. The genes are:
ND1, ND2, ND3, ND4, ND4L, ND5, ND6, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS5, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NDUFV3, NDUFA1, NDUFA2, NDUFA3, NDUFA4, NDUFA4L2, NDUFA5, NDUFA6, NDUFA7, NDUFA8, NDUFA9, NDUFA10, NDUFAB1, NDUFA11, NDUFA12, NDUFA13, NDUFB1, NDUFB2, NDUFB3, NDUFB4, NDUFB5, NDUFB6, NDUFB7, NDUFB8, NDUFB9, NDUFB10, NDUFB11, NDUFC1, NDUFC2, NDUFC2-KCTD14, SDHA, SDHB, SDHC, SDHD, UQCRFS1, CYTB, CYC1, UQCRC1, UQCRC2, UQCRH, UQCRHL, UQCRB, UQCRQ, UQCR10, UQCR11, COX10, COX3, COX1, COX2, COX4I2, COX4I1, COX5A, COX5B, COX6A1, COX6A2, COX6B1, COX6B2, COX6C, COX7A1, COX7A2, COX7A2L, COX7B, COX7B2, COX7C, COX8C, COX8A, COX11, COX15, COX17, CYCS, ATP5F1A, ATP5F1B, ATP5F1C, ATP5F1D, ATP5F1E, ATP5PO, ATP6, ATP5PB, ATP5MC1, ATP5MC2, ATP5MC3, ATP5PD, ATP5ME, ATP5MF, ATP5MG, ATP5PF, ATP8, ATP6V1A, ATP6V1B1, ATP6V1B2, ATP6V1C2, ATP6V1C1, ATP6V1D, ATP6V1E2, ATP6V1E1, ATP6V1F, ATP6V1G1, ATP6V1G3, ATP6V1G2, ATP6V1H, TCIRG1, ATP6V0A2, ATP6V0A4, ATP6V0A1, ATP6V0C, ATP6V0B, ATP6V0D1, ATP6V0D2, ATP6V0E1, ATP6V0E2, ATP6AP1, ATP4A, ATP4B, ATP12A, PPA2, PPA1, LHPP물량 쩌네.
Appendix. 뭐 딴거 없어요? 메인페이지에 개많던데?
나도 궁금해서 KEGG 메인에 있는거 다 돌려봤음.
from Bio.KEGG import REST
human_pathways = REST.kegg_list('brite').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('pathway').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('module').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('enzyme').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('genome').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('glycan').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('compound').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('reaction').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('disease').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('drug').read()
print(human_pathways)from Bio.KEGG import REST
human_pathways = REST.kegg_list('network').read()
print(human_pathways)저기서 되는거 Genes빼고 어지간한건 다 된다.
1. KEGG Brite
2. KEGG Pathway
3. KEGG Module
4. KEGG Enzyme
5. KEGG Compound
6. KEGG Genome Gene은 안되는데 Genome은 되네?
7. KEGG Glycan
8. KEGG Reaction
9. KEGG Network
10. KEGG Drug
11. KEGG Disease
Appendix 2. 응용편
from Bio.KEGG import REST
Disease = REST.kegg_list('disease').read()
# 가져올 수 있는 것: brite, pathway, genome(gene은 안됨), module, enzyme, glycan, compound, reaction, network, drug, disease
disease_leukemia = []
for line in Disease.rstrip().split("\n"):
entry, description = line.split("\t")
if "leukemia" in description:
disease_leukemia.append(description)
print(disease_leukemia)다 불러올 수 있으면 아까 그 쿼링 코드 써서 검색도 되나 궁금해서 찾아봤음.
from Bio.KEGG import REST
drug = REST.kegg_list('drug').read()
# 가져올 수 있는 것: brite, pathway, genome(gene은 안됨), module, enzyme, glycan, compound, reaction, network, drug, disease
drug_list = []
drug_name=input("찾고 싶은 약과 관련된 것을 입력해주세요: ")
for line in drug.rstrip().split("\n"):
entry, description = line.split("\t")
if drug_name in description:
drug_list.append(description)
print(drug_list)이건 나름 업그레이드 버전인데 Drug에서 사용자가 입력한 항목을 찾아준다. (와일드카드는 안 먹는다)
'Coding > Python' 카테고리의 다른 글
| 심심해서 써보는 본인 개발환경 (0) | 2022.08.21 |
|---|---|
| Biopython-Q&A (0) | 2022.08.21 |
| 텍스트 입력받아서 Wordcloud 만들기 (0) | 2022.08.21 |
| Biopython-Entrez에서 논문 제목 긁어와서 Wordcloud 만들기 (0) | 2022.08.21 |
| Biopython-Clustering 입력 인자 (0) | 2022.08.21 |

