와! 드디어 실전인가요? 근데 실전 생각보다 노잼임...
Distance matrix
거리 행렬. 두 점간의 거리를 배열해 행렬로 나타낸 것이다. 점이 N개일 때 Distance matrix는 N*N으로 표기할 수 있다.
import numpy as np
import pandas as pd
from Bio.Cluster import distancematrix
data=np.array([[0, 1, 2, 3],[4, 5, 6, 7],[8, 9, 10, 11],[1, 2, 3, 4]])
matrix = distancematrix(data)
# 뭐야 이거 왜 한영키 안먹어요
distances = distancematrix(data, dist='e')
print(distances)[array([], dtype=float64), array([16.]), array([64., 16.]), array([ 1., 9., 49.])]뭐여 행렬 어디갔어요!!! 아 일단 나도 저 반응이어서 이해는 함... 근데 Distance matrix의 성질을 잘 이용하면 저걸 행렬로 만들 수 있다.

여기서 이용할 Distance matrix의 성질 두 개는
1. Distance matrix도 인접행렬처럼 주대각선(좌상-우하)이 0이다. 다 0이다.
2. Distance matrix는 주대각선을 축으로 하는 대칭 행렬(대칭 행렬: 전치행렬이 원 행렬과 같음)이다.
이거다. 이걸 이용해 저 array를 좌측 하단에 채우고, 대칭되는 방향으로 우측을 채우면 된다. 얘네 이거 안 알려줘서 내가 찾음.
k- 들어가는 군집화
k-mean, k-median
from Bio.Cluster import kcluster
import numpy as np
data=np.array([[0,1,2,3],[4,5,6,7],[8,9,10,11],[0,1,2,3]])
clusterid, error, nfound = kcluster(data)
print("Clusterid:",clusterid, "error:",error, "nfound:",nfound)Clusterid: [0 1 1 0] error: 8.0 nfound: 1코드가 동일한데, method='a'(기본값)는 평균, method='m'은 중앙값. 위 코드는 평균으로 구한 것.
k-medoid
from Bio.Cluster import kmedoids
from Bio.Cluster import distancematrix
import numpy as np
data=np.array([[0, 1, 2, 3],[4, 5, 6, 7],[8, 9, 10, 11],[1, 2, 3, 4]])
matrix = distancematrix(data)
# 뭐야 이거 왜 한영키 안먹어요
distances = distancematrix(data, dist='e')
clusterid, error, nfound = kmedoids(distances)
print("clusterid:",clusterid,"error:",error,"nfound:",nfound)clusterid: [0 1 1 0] error: 17.0 nfound: 1얘는 배열이 아니라 거리 행렬을 입력으로 받는다. 아까 그 괴랄한 형태도 되고, 1차원이나 2차원 배열로도 받는다.
Hierarchical clustering
method
거리를 재는 방식에 따라 나뉜다. 거리 함수랑 별개.
Pairwise single-linkage clustering: 제일 짧은 거리(s)
Pairwise maximum-linkage clustering: 제일 긴 거리(m)
Pairwise average-linkage clustering: 평균 거리(a)
Pairwise centroid-linkage clustering: 무게중심(centroid, c)
노드 생성
from Bio.Cluster import Node
A=Node(2,3) # left, right만 존재(거리=0)하는 노드
B=Node(2,3,0.91) # left, right, distance가 셋 다 존재하는 노드
print(B.left,B.right,B.distance)2 3 0.91입력인자는 왼쪽, 오른쪽, 거리. 거리는 option이라 입력 안 해도 된다.
from Bio.Cluster import Node, Tree
nodes = [Node(1, 2, 0.2), Node(0, 3, 0.5), Node(-2, 4, 0.6), Node(-1, -3, 0.9)]
tree = Tree(nodes)
print(tree)(1, 2): 0.2
(0, 3): 0.5
(-2, 4): 0.6
(-1, -3): 0.9이런 식으로 트리를 그릴 수도 있다. 물론 Tree 모듈은 모셔와야 한다.
print(tree[0])
# 생성된 tree에서 indexing을 통해 노드에 접근할 수 있다.
print(tree[0].left)
# 노드에 접근해서 노드의 정보를 가져올 수도 있다.(1, 2): 0.2
1인덱싱을 할 수도 있다. 트리의 노드를 인덱싱하거나, 아래 코드처럼 트리의 노드에 대한 정보(왼쪽, 오른쪽, 거리)를 가져오는 것도 된다.
indices = tree.sort()
clusterid = tree.cut(nclusters=2)
print("indices: ",indices)
print("clusterid: ",clusterid)
# 누구 저 sort 어떻게 쓰는 지 아는 사람?indices: [7 0 1 2 1]
clusterid: [1 1 1 1 1]정렬이랑 분할(cut이 분할)도 된다는데 sort 저거 옵션 어떻게 주는건지는 안나왔다.
본격적으로 해보기
from Bio.Cluster import treecluster
import numpy as np
data=np.array([[1,2,3,4],[5,6,7,8],[9,10,11,12],[0,1,2,3]])
tree = treecluster(data)
print(tree)(3, 0): 1
(2, 1): 16
(-2, -1): 81Array로 clustering한 결과.
tree = treecluster(data,dist="b",distancematrix=None)
print(tree)
# 다른 옵션을 줄 수도 있다.(3, 0): 1
(2, 1): 4
(-2, -1): 9거리 함수 옵션을 유클리드 거리에서 시티 블록으로 바꿨다.
distances=distancematrix((data))
tree = treecluster(data=None,distancematrix=distances)
print(tree)
# Distance matrix를 미리 계산해 그걸로 그릴 수도 있다.
# ValueError: use either data or distancematrix; do not use both
# Data와 Distance matrix중 하나는 None이어야 한다. 안그러면 위 에러가 반긴다.(3, 0): 1
(2, 1): 16
(-2, -1): 81같은 array로 그린건데 arrya가 아니라 거리 행렬을 구해서 그린 것. Data나 distancematrix는 둘 중 하나만 들어갈 수 있다. 그리고 Distance matrix로 hierarchical clustering을 할 때는 거리 측정 method 중 centroid(‘c’)를 선택할 수 없다.
Self-Organizing Maps
대뇌피질의 시각피질을 모델화했다나… 모델화 한 사람 이름이 Teuvo Kohenen이라 Kohenen map이라고도 부른다. k-mean에 위상 개념이 추가된 것.

Adjustment: 보정치(벡터에서 클러스터 찾을 때 쓰는 값
Radius: 반경
그렇다고 한다. 나도 잘 모름.
from Bio.Cluster import somcluster
import numpy as np
data=np.array([[1,2,3,4],[5,6,7,8],[9,10,11,12],[0,1,2,3]])
clusterid, celldata = somcluster(data)
print("clusterid:",clusterid,"\n","celldata:",celldata)clusterid: [[1 0]
[1 0]
[1 0]
[1 0]]
celldata: [[[-1.80107153 -0.6130221 0.16768853 0.59348614]]
[[ 1.6816792 -0.16877007 -0.34863638 1.01090276]]]
Principal Component Analysis
주성분 분석. 고차원의 데이터를 저차원으로 축소시키는 기법 중 하나라고 한다. 아니 무슨 선형대수학이 왜 나오는거냐 대체... 와 진짜 대환장 파티네 뭐 주성분의 표본을 직교변환한다던가... 이런건 나도 모르겠고... 저는 수학이랑 담 쌓아서요... I HATE MATH 쌉가능
from Bio.Cluster import pca
import numpy as np
data=np.array([[1,2,3,4],[5,6,7,8],[9,10,11,12],[0,1,2,3]])
columnmean, coordinates, components, eigenvalues = pca(data)
print("columnmean:",columnmean)
print("coordinates:",coordinates)
print("components:",components)
print("eigenvalues:",eigenvalues)columnmean: [3.75 4.75 5.75 6.75]
coordinates: [[ 5.50000000e+00 -6.00596572e-16 -2.63550106e-49 -0.00000000e+00]
[-2.50000000e+00 -1.16262943e-16 3.00734525e-47 0.00000000e+00]
[-1.05000000e+01 -4.49845107e-16 -7.36989119e-48 0.00000000e+00]
[ 7.50000000e+00 -2.28099977e-16 -1.00093434e-49 0.00000000e+00]]
components: [[-5.00000000e-01 -5.00000000e-01 -5.00000000e-01 -5.00000000e-01]
[ 8.66025404e-01 -2.88675135e-01 -2.88675135e-01 -2.88675135e-01]
[ 0.00000000e+00 8.16496581e-01 -4.08248290e-01 -4.08248290e-01]
[ 0.00000000e+00 -7.19816116e-17 -7.07106781e-01 7.07106781e-01]]
eigenvalues: [1.42478068e+01 7.92857827e-16 3.09646140e-47 0.00000000e+00]여기 표시된 결과는 대충 이렇다.
Columnmean: 컬럼 평균(어레이 가로 말고 세로)
Coordinates: 주성분에 대한 데이터의 행 좌표
Components: 주성분
Eigenvalues: 각 주성분의 고유값
Cluster/TreeView 파일 핸들링하기
예제파일... 얘네가 깃헙에 올려뒀으니까 받아가세여...
from Bio import Cluster
with open("/home/koreanraichu/cyano.txt") as handle:
record = Cluster.read(handle)
print(record)<Bio.Cluster.Record object at 0x7ff32bb85af0>불러와서 레코드만 달랑 달라고 하면 저렇게 준다. record.geneid 이런 식으로 세부 요소들을 요청하자.
from Bio import Cluster
with open("/home/koreanraichu/cyano.txt") as handle:
record = Cluster.read(handle)
# 불러왔다
matrix = record.distancematrix()
# Distance matrix 계산
cdata, cmask = record.clustercentroids()
# Cluster 무게중심(Centroid) 계산
distance = record.clusterdistance()
# 클러스터간 거리 계산
tree = record.treecluster()
# hierarchical clustering
# 이거 matplot으로 못뽑나...
clusterid, error, nfound = record.kcluster()
# k-mean clustering
# method='a': k-mean
# method='m': k-median
clusterid, celldata = record.somcluster()
# SOM 계산하기
jobname="cyano_clustering"
record.save(jobname, record.treecluster(), record.treecluster(transpose=1))
# 내 컴퓨터에 저(별)장
# 기본 형식: record.save(jobname, geneclusters, expclusters)
# geneclusters=record.treecluster()
# expclusters=record.treecluster(transpose=1)본인 깃헙에 있는 전체 소스. 저장의 경우 jobname, geneclusters, expclusters에 입력하는 만큼 파일이 생긴다.
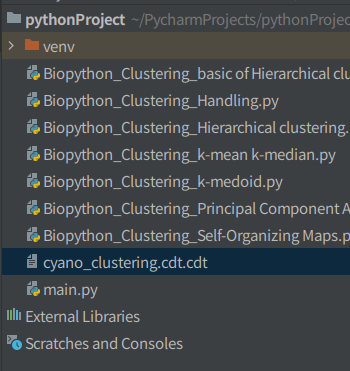
jobname만 입력하면 이거 하나 생긴다.
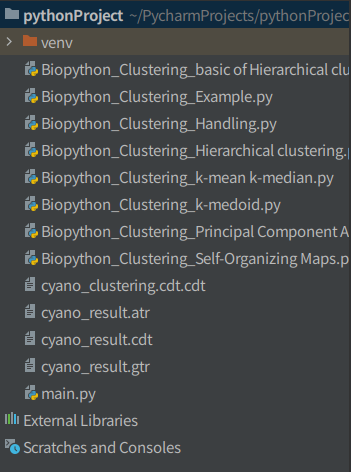
셋 다 입력하면 세개 생긴다.
from Bio import Cluster
with open("/home/koreanraichu/cyano.txt") as handle:
record = Cluster.read(handle)
# 불러와서
genetree = record.treecluster(method="s")
genetree.scale()
# Hierarchical clustering
# method는 Pairwise single-linkage clustering
exptree = record.treecluster(dist="u", transpose=1)
# 이것도 Hierarchical clustering(컬럼으로 그림, dist)
# dist는 Uncentered Pearson correlation(어슨이형)
# 그려서
record.save("cyano_result", genetree, exptree)
# 저장아무튼 이런 식으로 쓴다고...
'Coding > Python' 카테고리의 다른 글
| Biopython-Entrez에서 논문 제목 긁어와서 Wordcloud 만들기 (0) | 2022.08.21 |
|---|---|
| Biopython-Clustering 입력 인자 (0) | 2022.08.21 |
| Biopython으로 Clusting analysis 하기 (이론편) (0) | 2022.08.21 |
| Biopython으로 Sequence motif analysis 하기 (0) | 2022.08.21 |
| Biopython으로 Phylogenetic tree 그리기 (0) | 2022.08.21 |


