모티브 찾으면 구조 모티브가 나오는데 이거는 '단백질이나 핵산과 같은 사슬 모양의 생체 분자에서 진화적으로 관련이 없는 다양한 분자에서 나타나는 일반적인 3차원 구조'로 정의한다. 근데 여기서 다루는 모티브는 그거 말고 시퀀스 모티브...

대충 이런거다. 위 그림은 뭔 시퀀스인지는 모르겠으나 3, 4, 5번째 염기가 GAA가 압도적으로 많은 듯.
오늘은 대충
from Bio import motifs이런거 부른다.
Motif object
객체 생성하기
from Bio import motifs
from Bio.Seq import Seq
instances=[Seq("TGTCGTATCG"),Seq("GTAAATAGCC"),Seq("GTAAATAACC"),Seq("TCGCGGAGCC"),Seq("ATGTGCCATA"),Seq("CTCGTCTGCG")]
m = motifs.create(instances)
print(m)TGTCGTATCG
GTAAATAGCC
GTAAATAACC
TCGCGGAGCC
ATGTGCCATA
CTCGTCTGCG이런 식으로 시퀀스 객체를 활용해서 만들 수 있다. 저거는 DNA 랜덤으로 만든거. len() 쓰면 길이도 알려준다.
print(m.counts) 0 1 2 3 4 5 6 7 8 9
A: 1.00 0.00 2.00 2.00 2.00 0.00 4.00 2.00 0.00 1.00
C: 1.00 1.00 1.00 2.00 0.00 2.00 1.00 0.00 5.00 3.00
G: 2.00 1.00 2.00 1.00 3.00 1.00 0.00 3.00 0.00 2.00
T: 2.00 4.00 1.00 1.00 1.00 3.00 1.00 1.00 1.00 0.00저 표가 뭔 소리냐면... 일단 저 DNA가 10bp짜리 6개 시퀀스로 만든건데(그래서 0부터 9까지), 6개 시퀀스의 0번 컬럼을 다 봤더니 아데닌과 시토신이 있는 게 하나, 구아닌과 티민이 있는 게 두개라는 얘기.
print(m.counts["A"])[1, 0, 2, 2, 2, 0, 4, 2, 0, 1]인덱싱은 넘파이 array처럼 2차원으로 할 수 있다. 저건 염기(인덱스)로 뽑은거.
print(m.counts["A",0])1첫번째 염기에 아데닌으로 뽑은 거.
print(m.counts[:,0]){'A': 1, 'C': 1, 'G': 2, 'T': 2}컬럼 인덱싱도 된다.
print(m.consensus)
print(m.anticonsensus)GTAAGTAGCC
AACGCAGCAT순서대로
-가장 많은 염기
-가장 적은 염기
를 표시해준다.
print(m.degenerate_consensus)NTNNRYARCS...... 어머니... 이게 무슨 개소리죠...

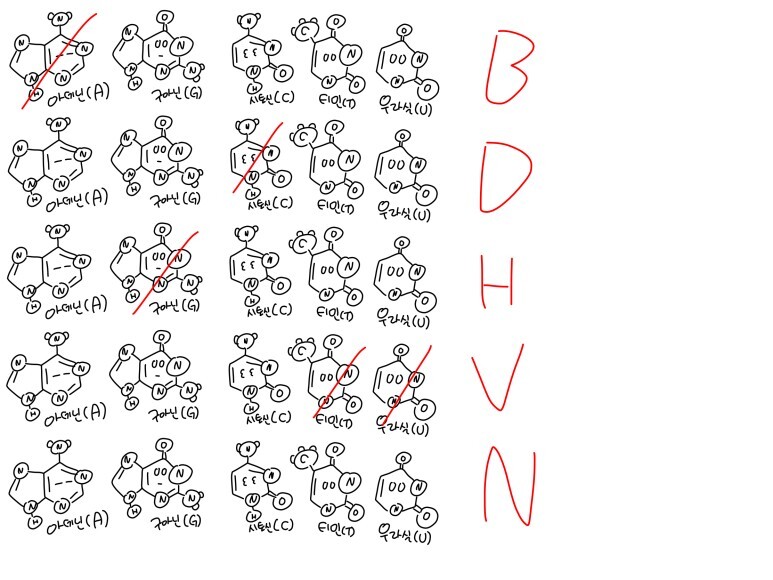
당황하지 말고 이걸 보자.

이것도 있다. (FASTA 문서에 있음)
그니까 저기 다섯번째에 있는 R은 '아 이거 내가 셔봤는데 대충 퓨린이 많어' 이런 얘기.
r = m.reverse_complement()
print(r)CGATACGACA
GGCTATTTAC
GGTTATTTAC
GGCTCCGCGA
TATGGCACAT
CGCAGACGAGreverse complement 만들어서 셀 수도 있다.
Weblogo 만들기
m.weblogo("mymotif.png")
님아 사이즈좀...
Data 읽기
이 와중에 이것도 DB가 있다. JASPAR라고...
https://jaspar.genereg.net/
JASPAR: An open-access database of transcription factor binding profiles
JASPAR is the largest open-access database of curated and non-redundant transcription factor (TF) binding profiles from six different taxonomic groups.
jaspar.genereg.net
여기다.
sites format은 못구했고 jaspar format이랑 pfm 읽어봅시다. meme이랑 tr뭐시기는 에러났음.
from Bio import motifs
fh = open("/home/koreanraichu/MA0004.1.jaspar")
for m in motifs.parse(fh, "jaspar"):
print(m)TF name Arnt
Matrix ID MA0004.1
Matrix:
0 1 2 3 4 5
A: 4.00 19.00 0.00 0.00 0.00 0.00
C: 16.00 0.00 20.00 0.00 0.00 0.00
G: 0.00 1.00 0.00 20.00 0.00 20.00
T: 0.00 0.00 0.00 0.00 20.00 0.00jaspar format
from Bio import motifs
with open("Arnt.sites") as handle:
arnt = motifs.read(handle, "sites")sites format은 이렇게 부른다.
with open("/home/koreanraichu/MA0007.1.pfm") as handle:
srf = motifs.read(handle, "pfm")
print(srf)pfm은 이렇게 부르면 되는데 문제가 하나있다.

저 위에 있는 ID를 인식을 못 해서 저거 저대로 부르면 ValueError: could not convert string to float: '>MA0007.1' 이 여러분을 반긴다.
TF name None
Matrix ID None
Matrix:
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
A: 9.00 9.00 11.00 16.00 0.00 12.00 21.00 0.00 15.00 4.00 5.00 6.00 3.00 0.00 4.00 11.00 1.00 3.00 6.00 6.00 10.00 5.00
C: 7.00 2.00 3.00 1.00 0.00 6.00 2.00 24.00 0.00 9.00 11.00 9.00 5.00 0.00 0.00 5.00 22.00 16.00 7.00 5.00 11.00 11.00
G: 2.00 3.00 3.00 7.00 22.00 1.00 0.00 0.00 8.00 2.00 2.00 9.00 1.00 24.00 0.00 1.00 1.00 1.00 5.00 9.00 0.00 6.00
T: 6.00 10.00 7.00 0.00 2.00 5.00 1.00 0.00 1.00 9.00 6.00 0.00 15.00 0.00 20.00 7.00 0.00 4.00 6.00 4.00 3.00 2.00ID 지우면 이렇게 부를 수는 있는데 그럴거면 불러서 뭐함?
from Bio import motifs
with open("/home/koreanraichu/MA0273.1.meme") as handle:
record = motifs.parse(handle, "meme")
print(record)Meme은 이렇게 부르는데

이게 이 형식이그덩... 그랬더니 ValueError: Improper MEME XML input file. XML root tag should start with <MEME version= ... 뜸...
from Bio import motifs
with open("/home/koreanraichu/MA0273.1.transfac") as handle:
record = motifs.parse(handle, "TRANSFAC")
print(record)transfac은 이렇게 부르는데

얘는 또 ValueError: A TRANSFAC motif line should have 2 spaces between key and value columns: "AC MA0273.1"가 반긴다. 니네 뭐하자는...
Data 쓰기
from Bio import motifs
fh = open("/home/koreanraichu/MA0004.1.jaspar")
for m in motifs.parse(fh, "jaspar"):
print(m.format("pfm")) 4.00 19.00 0.00 0.00 0.00 0.00
16.00 0.00 20.00 0.00 0.00 0.00
0.00 1.00 0.00 20.00 0.00 20.00
0.00 0.00 0.00 0.00 20.00 0.00Jaspar->pfm
from Bio import motifs
fh = open("/home/koreanraichu/MA0004.1.jaspar")
for m in motifs.parse(fh, "jaspar"):
print(m.format("transfac"))P0 A C G T
01 4 16 0 0 C
02 19 0 1 0 A
03 0 20 0 0 C
04 0 0 20 0 G
05 0 0 0 20 T
06 0 0 20 0 G
XX
//Jaspar->transfac (meme은 지원 안함)
instances=[Seq("TGTCGTATCG"),Seq("GTAAATAGCC"),Seq("GTAAATAACC"),Seq("TCGCGGAGCC"),Seq("ATGTGCCATA"),Seq("CTCGTCTGCG")]
m2 = motifs.create(instances)
print(m2.format("jaspar"))>None
A [ 1.00 0.00 2.00 2.00 2.00 0.00 4.00 2.00 0.00 1.00]
C [ 1.00 1.00 1.00 2.00 0.00 2.00 1.00 0.00 5.00 3.00]
G [ 2.00 1.00 2.00 1.00 3.00 1.00 0.00 3.00 0.00 2.00]
T [ 2.00 4.00 1.00 1.00 1.00 3.00 1.00 1.00 1.00 0.00]물론 jaspar format으로도 된다.
PWM(position weight matrices)
pwm = m.counts.normalize(pseudocounts=0.5)
print(pwm) 0 1 2 3 4 5
A: 0.20 0.89 0.02 0.02 0.02 0.02
C: 0.75 0.02 0.93 0.02 0.02 0.02
G: 0.02 0.07 0.02 0.93 0.02 0.93
T: 0.02 0.02 0.02 0.02 0.93 0.02normalize가 들어간 걸 보면 정규화를 하는 모양. 정규화나 표준화는 원래 인공지능용 Data 전처리 과정에서 진행한다.
pwm = m.counts.normalize(pseudocounts={"A":0.6, "C": 0.4, "G": 0.4, "T": 0.6})
print(pwm) 0 1 2 3 4 5
A: 0.21 0.89 0.03 0.03 0.03 0.03
C: 0.75 0.02 0.93 0.02 0.02 0.02
G: 0.02 0.06 0.02 0.93 0.02 0.93
T: 0.03 0.03 0.03 0.03 0.94 0.03염기별로 pseudocount를 지정할 수도 있다.
rpwm = pwm.reverse_complement()
print(rpwm)
# reverse complement로도 된다. (아마 밑에 코드로 계산된 듯) 0 1 2 3 4 5
A: 0.03 0.94 0.03 0.03 0.03 0.03
C: 0.93 0.02 0.93 0.02 0.06 0.02
G: 0.02 0.02 0.02 0.93 0.02 0.75
T: 0.03 0.03 0.03 0.03 0.89 0.21reverse complement로도 된다.
print(pwm.consensus)
print(pwm.anticonsensus)
print(pwm.degenerate_consensus)
# 제일 많은거/제일 적은거/Degenerate consensusCACGTG
GCGCCC
CACGTG위에 세 개를 저걸로 결론내는 모양. 아 그래서 2:2인데 아데닌 나왔냐
PSSM(position specific scoring matrices)
이거는 PWM이 있어야 구할 수 있는 모양.
pwm = m.counts.normalize(pseudocounts=0.5) # 예제 코드를 보니 얘가 있어야 계산할 수 있는 모양
pssm = pwm.log_odds()
print(pssm) 0 1 2 3 4 5
A: -0.29 1.83 -3.46 -3.46 -3.46 -3.46
C: 1.58 -3.46 1.90 -3.46 -3.46 -3.46
G: -3.46 -1.87 -3.46 1.90 -3.46 1.90
T: -3.46 -3.46 -3.46 -3.46 1.90 -3.46저기서 양수면 모티브에서, 음수면 백그라운드에서 많이 보인다. 백그라운드에서 많이 보인다는 건 '주'는 아닌 듯... 0이면? 또이또이 쌤쌤.
background = {"A":0.3,"C":0.2,"G":0.2,"T":0.3}
pssm = pwm.log_odds(background)
print(pssm)
# background를 염기별로 줄 수도 있다. 0 1 2 3 4 5
A: -0.55 1.56 -3.72 -3.72 -3.72 -3.72
C: 1.91 -3.14 2.22 -3.14 -3.14 -3.14
G: -3.14 -1.55 -3.14 2.22 -3.14 2.22
T: -3.72 -3.72 -3.72 -3.72 1.64 -3.72염기별로 background setting도 된다. 최대/최소값을 볼 수 있고, background를 임의로 부여한 경우 평균과 표준편차도 구할 수 있다.
print("%4.2f" % pssm.max) # 최대값
print("%4.2f" % pssm.min) # 최소값
mean = pssm.mean(background) # 평균
std = pssm.std(background) # 표준편차
print("mean = %0.2f, standard deviation = %0.2f" % (mean, std))
그 이름 검⭐색
시퀀스로 찾기
from Bio import motifs
from Bio.Seq import Seq
test_seq=Seq("CTCGTCTGCG")
m = motifs.create(instances=[Seq("TGTCGTATCG"),Seq("GTAAATAGCC"),Seq("GTAAATAACC"),Seq("TCGCGGAGCC"),Seq("ATGTGCCATA"),Seq("CTCGTCTGCG")])
for pos, seq in m.instances.search(test_seq):
print(pos,seq)0 CTCGTCTGCGtest_seq에서 motif를 찾는거기때문에 test_seq보다 motif가 더 길면 에러난다.
PSSM score로 찾기
pwm = m.counts.normalize(pseudocounts=0.5) # 예제 코드를 보니 얘가 있어야 계산할 수 있는 모양
pssm = pwm.log_odds()
print(pssm)
# PSSM으로 찾기(PSSM 도출)
for position, score in pssm.search(test_seq, threshold=3.0):
print("Position %d: score = %5.3f" % (position, score))
# 찾았음!Position 0: score = 2.005threshold요? 그거 밑에 나옴. 아마 3.0이 기준인 듯 하다.
Threshold
background = {"A":0.3,"C":0.2,"G":0.2,"T":0.3}
distribution = pssm.distribution(background=background, precision=10**4)
threshold = distribution.threshold_fpr(0.01)
print("%5.3f" % threshold)3.784있으니까 하긴 한다만... 이거 뭐예여...
threshold = distribution.threshold_fnr(0.1)
print("%5.3f" % threshold)-0.664일단 위에껀 fpr 밑에껀 fnr이다.
-fpr: false-positive rate (찾을 확률)
-fnr: false-negative rate (못찾을 확률)
-balanced: false-positive rate and the false-negative rate (fnr/fpr≃ t)
-patser: equality between the −log of the false-positive rate and the information content
이렇다고 한다. 저걸로 threshold 계산해서 그거 때려박고 검색할 수도 있다.
background = {"A":0.3,"C":0.2,"G":0.2,"T":0.3}
distribution = pssm.distribution(background=background, precision=10**4)
threshold = distribution.threshold_fnr(0.1)
print("%5.3f" % threshold)
for position, score in pssm.search(test_seq, threshold=threshold):
print("Position %d: score = %5.3f" % (position, score))Position 0: score = 6.703
Position -22: score = 0.084
Position 5: score = -0.301
Position 9: score = 0.195
Position 12: score = -0.346
Position -12: score = 0.681
Position 15: score = 2.992이게 threshold 계산해서 때려박은 결과
Position 0: score = 6.703
Position -22: score = 0.084
Position 9: score = 0.195
Position -12: score = 0.681
Position 15: score = 2.992이건 똑같은 모티브에서 고정값threshold로 찾은 결과이다.
PSSM과 motif
print(motif.counts)
print(motif.pwm)
print(motif.pssm)
# 일단 뽑고 보는 타입0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
A: 64.00 104.00 484.00 78.00 455.00 248.00 98.00 52.00 348.00 12.00 966.00 26.00 9.00 47.00 892.00 487.00 288.00 373.00 178.00 402.00 435.00
C: 217.00 552.00 111.00 401.00 51.00 34.00 724.00 918.00 346.00 3.00 5.00 6.00 10.00 930.00 42.00 123.00 140.00 110.00 367.00 140.00 229.00
G: 425.00 150.00 114.00 186.00 370.00 670.00 143.00 7.00 280.00 977.00 23.00 962.00 4.00 7.00 16.00 320.00 317.00 81.00 184.00 341.00 241.00
T: 292.00 192.00 288.00 333.00 122.00 46.00 33.00 20.00 24.00 6.00 4.00 4.00 975.00 15.00 49.00 68.00 254.00 434.00 268.00 114.00 94.00
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
A: 0.06 0.10 0.49 0.08 0.46 0.25 0.10 0.05 0.35 0.01 0.97 0.03 0.01 0.05 0.89 0.49 0.29 0.37 0.18 0.40 0.44
C: 0.22 0.55 0.11 0.40 0.05 0.03 0.73 0.92 0.35 0.00 0.01 0.01 0.01 0.93 0.04 0.12 0.14 0.11 0.37 0.14 0.23
G: 0.43 0.15 0.11 0.19 0.37 0.67 0.14 0.01 0.28 0.98 0.02 0.96 0.00 0.01 0.02 0.32 0.32 0.08 0.18 0.34 0.24
T: 0.29 0.19 0.29 0.33 0.12 0.05 0.03 0.02 0.02 0.01 0.00 0.00 0.98 0.02 0.05 0.07 0.25 0.43 0.27 0.11 0.09
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
A: -1.96 -1.26 0.96 -1.68 0.87 -0.01 -1.35 -2.26 0.48 -4.38 1.95 -3.26 -4.79 -2.41 1.84 0.96 0.21 0.58 -0.49 0.69 0.80
C: -0.20 1.15 -1.17 0.68 -2.29 -2.88 1.54 1.88 0.47 -6.38 -5.64 -5.38 -4.64 1.90 -2.57 -1.02 -0.84 -1.18 0.56 -0.83 -0.13
G: 0.77 -0.73 -1.13 -0.42 0.57 1.43 -0.80 -5.15 0.17 1.97 -3.44 1.95 -5.96 -5.16 -3.96 0.36 0.34 -1.62 -0.44 0.45 -0.05
T: 0.23 -0.38 0.21 0.42 -1.03 -2.44 -2.92 -3.64 -3.38 -5.38 -5.96 -5.96 1.97 -4.06 -2.35 -1.88 0.02 0.80 0.10 -1.13 -1.41일단 불러서 뽑아보면 이런 식으로 나온다. (순서대로 count, PWM, PSSM)
print(motif.background)
print(motif.pseudocounts)
# 일단 불러온 데이터에서 뽑을 수는 있다.{'A': 0.25, 'C': 0.25, 'G': 0.25, 'T': 0.25}
{'A': 0.0, 'C': 0.0, 'G': 0.0, 'T': 0.0}Motif 데이터에 손대지 않은 default값.
motif.pseudocounts = 3.0
motif.background = {"A": 0.2, "C": 0.3, "G": 0.3, "T": 0.2}
# 어? 되네?
print(motif.counts)
print(motif.pwm)
print(motif.pssm)
# 바꾸고 계산해서 보자 한번.
print(motif.background)
print(motif.pseudocounts)0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
A: 0.07 0.11 0.48 0.08 0.45 0.25 0.10 0.05 0.35 0.01 0.96 0.03 0.01 0.05 0.89 0.49 0.29 0.37 0.18 0.40 0.43
C: 0.22 0.55 0.11 0.40 0.05 0.04 0.72 0.91 0.35 0.01 0.01 0.01 0.01 0.92 0.04 0.12 0.14 0.11 0.37 0.14 0.23
G: 0.42 0.15 0.12 0.19 0.37 0.67 0.14 0.01 0.28 0.97 0.03 0.96 0.01 0.01 0.02 0.32 0.32 0.08 0.19 0.34 0.24
T: 0.29 0.19 0.29 0.33 0.12 0.05 0.04 0.02 0.03 0.01 0.01 0.01 0.97 0.02 0.05 0.07 0.25 0.43 0.27 0.12 0.10
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
A: -1.59 -0.92 1.27 -1.32 1.18 0.31 -1.00 -1.88 0.80 -3.75 2.26 -2.80 -4.07 -2.02 2.15 1.28 0.53 0.90 -0.16 1.00 1.12
C: -0.46 0.87 -1.41 0.42 -2.49 -3.03 1.26 1.61 0.20 -5.66 -5.24 -5.07 -4.54 1.62 -2.75 -1.27 -1.08 -1.42 0.29 -1.08 -0.39
G: 0.50 -0.99 -1.37 -0.68 0.30 1.15 -1.05 -4.92 -0.10 1.69 -3.54 1.67 -5.44 -4.92 -4.00 0.09 0.08 -1.85 -0.69 0.18 -0.31
T: 0.55 -0.05 0.53 0.73 -0.69 -2.04 -2.49 -3.13 -2.90 -4.49 -4.85 -4.85 2.28 -3.49 -1.96 -1.51 0.35 1.11 0.43 -0.79 -1.06
{'A': 0.2, 'C': 0.3, 'G': 0.3, 'T': 0.2}
{'A': 3.0, 'C': 3.0, 'G': 3.0, 'T': 3.0}아, 가져온 데이터에서 바꿔줄 수는 있다. 저게 해본거임.
motif.background = None
print(motif.background)
# None: 각 염기별로 0.25씩
motif.background = 0.9
print(motif.background)
# GC 비중보게.
# {'A': 0.04999999999999999, 'C': 0.45, 'G': 0.45, 'T': 0.04999999999999999} 는 심한 거 아니냐 근데.뭐 이렇게 쓰기도 한다고...
저 값을 바꿔주게 되면 당연히 평균, 표준편차, Threshold도 바뀐다.
Motif 비교하기
참고로 이거 일단 두 개 불렀다.
motif_1.pseudocounts = {"A":0.6, "C": 0.4, "G": 0.4, "T": 0.6}
motif_1.background = {"A":0.3,"C":0.2,"G":0.2,"T":0.3}
# pseudocount와 background를 설정해준다(깔맞춤)
pssm_motif_1=motif_1.pssm
print(pssm_motif_1)
# pssm을 생성한다0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
A: 0.78 -0.06 -0.33 -1.50 1.55 1.66 1.66 -1.27 -1.30 -2.21 1.59 1.62 1.59 -0.82 -0.40
C: -0.57 0.33 -1.28 -2.30 -3.11 -3.63 -4.15 1.65 -1.74 -3.31 -3.39 -3.15 -3.10 0.69 0.39
G: -0.51 0.30 -0.62 2.02 -1.61 -3.60 -3.70 0.08 -3.51 2.16 -2.01 -2.65 -2.36 0.94 -0.70
T: -0.52 -0.48 0.87 -2.83 -3.28 -3.81 -3.76 -3.06 1.42 -3.98 -3.47 -3.69 -3.24 -1.28 0.37귀찮아서 앞쪽 코드를 생략하긴 했는데 jaspar 파일을 두 개 불러서 background와 pesudocount를 하나로 맞추고 PSSM을 도출했다. 그래야 비교가 가능하다.
distance, offset = pssm_motif_2.dist_pearson(pssm_motif_1)
print("distance = %5.3g" % distance)
print("offset: ",offset)distance = 0.286
offset: -14저거 예제 코드에 pssm.dist_pearson으로 되어 있던거 걍 복붙했더니 에러나던데? 그런 이름 없셈! 을 토하더라… 참고로 저 코드에 같은 pssm이 들어가면 당연하게도 distance나 offset이 0이 된다.
'Coding > Python' 카테고리의 다른 글
| Biopython으로 Clusting analysis 하기 (실전편) (0) | 2022.08.21 |
|---|---|
| Biopython으로 Clusting analysis 하기 (이론편) (0) | 2022.08.21 |
| Biopython으로 Phylogenetic tree 그리기 (0) | 2022.08.21 |
| Biopython으로 PDB 탐방하기 (0) | 2022.08.21 |
| Biopython으로 Swiss-prot과 ExPASy 데이터베이스 탐방하기 (0) | 2022.08.21 |



