


MSA: multiple sequence alignment
여기에 관한 이론적인 설명은 나중에 또 입 털어드림 ㅇㅇ
아 참고로 MSA 관련해서 다른건 다 결과가 제대로 나왔는데 툴 관련해서 결과가 안나왔어요
이게 암만 찾아도 답이 없어서 좀 더 알아보고 수정할 가능성이 있음
시범조교 앞으로
오늘은 시범조교 가짓수가 좀 많은데 그 중에서도 FASTA 파일들을 좀 올리고자 한다. 이거 말고도 pfam에서 두갠가 받았는데 그건 걍 가서 암거나 받으면 된다.
해당 파일은 박테리아의 16s rRNA 시퀀스가 들어있는 파일이다. FASTA 파일이라 메모장 있으면 일단 열 수는 있다. 리눅스에서는 gedit으로 만들고 편집하고 다 했다. (vim 안씀) rRNA 시퀀스는 Silva에서 가져왔다. 고마워요 실바!
Agrobacterium
A. radiobacter를 필두로 하는 뿌리혹세균들(밑에 두놈도 뿌리에 혹 만드는지는 모름)
-Agrobacterium
Agrobacterium radiobacter (구 Agrobacterium tumefaciens)
Agrobacterium agile
Agrobacterium pusense
Agrobacterium salinitolerans
-Rhizobium
Rhizobium tropici (일반적으로 알고 있는 뿌리혹박테리아)
Rhizobium hainanense
Rhizobium gallicum
Rhizobium fabae
-Hoeflea
Hoeflea alexandrii
Hoeflea halophila 웬지 짠 거 좋아하게 생겼는데? 할로박테리움같은건가
Hoeflea trophica
-Ciceribacter
Ciceribacter lividus
Ciceribacter azotifigens
Ciceribacter thiooxidans
Enterobacter
E.coli를 필두로 하는 장내 세균들
-Enterococcus
Enterococcus faecalis (어디서 많이 봤음)
Enterococcus hirae
Enterococcus avium
Enterococcus caccae
-Escherichia
Escherichia coli (그냥 어디서나 볼 수 있는 대장균)
Escherichia coli O157:H7 (감염되면 X되는 대장균)
Escherichia albertii
Escherichia fergusonii
-Shigella
Shigella boydii
Shigella sonnei
Shigella dysenteriae
Shigella flexneri
Lactobacillus
님들 많이 드시는 그 유산균 맞습니다.
-Lactobacillus
Lactobacillus acidophilus
Lactobacillus helveticus
Lactobacillus plantarum (이분도 김치에서 발견된다)
Lactobacillus thailandensis DSM 22698 = JCM 13996 (아마 뒤에껀 strain 이름인 듯)
-Leuconostoc
Leuconostoc carnosum
Leuconostoc mesenteroides
Leuconostoc kimchii (이건 있다)
Leuconostoc miyukkimchii (저자 나와봐요 미역김치는 무슨 저세상 학명이야)
-Bifidobacterium (얘네는 방선균인데 일단 유산균으로 섭취 하기는 함... 비피더스 뭐 이런거)
Bifidobacterium bifidum
Bifidobacterium actinocoloniiforme DSM 22766 (아마 strain 이름...)
Bifidobacterium catenulatum
아니 근데 미역김치 학명 실화냐 대체 어디서 발견하면 학명이 저렇게 되는건데요 아니 저기 김치 하나 있는거 뭔데 김치요 참고로 C.kimchii(구 L.kimchii)는 데이터 없어서 못 넣었음... 김치 파티 각 나왔다
시퀀스 가져오기
MSA를 할래도 시퀀스를 가져와야 한다. 그것도 하나 말고 뭉텅이로. 여기서도 read()와 parse()로 나뉘긴 한데, 이거는 기존에 SeqIO로 읽을 때처럼 시퀀스 갯수가 여러 개 있어도 single alignment면 read()로 읽을 수 있다.
참고로 쿡북 예제로 스톡홀름 파일이 나오긴 했는데 FASTA도 불러올수는 있다.
from Bio import AlignIO
alignment = AlignIO.read("/home/koreanraichu/agrobacterium.fasta", "fasta")
print(alignment)MSA를 할 때는 SeqIO가 아니라 AlignIO로 불러오면 된다.
Alignment with 14 rows and 1561 columns
AUUCUCAACUUGAGAGUUUGAUCCUGGCUCAGAACGAACGCUGG...AAG AB102732.157.1651
................................AACGAACGCUGG...... AB680818.1.1406
................................AUUGAACGCUGG...... AB680959.1.1462
................................AACGAACGCUGG...... AB682466.1.1406
................................AACGAACGCUGG...... AB682468.1.1406
......................................CGCUGG...... AB969785.1.1417
............AGAGUUUGAUCCUGGCUCAGAACGAACGCUGG...... AJ786600.1.1449
.................................................. DQ835303.1.1353
.................................................. GU564401.1.1419
................................AACGAACGCUGG...... JF957616.1.1410
.................................................. JQ230000.1.1392
.................................................. KP142169.1.1341
............AGAGUUUGAUCAUGGCUCAGAACGAACGCUGG...... KU975391.1.1451
...............................A-ACGAACGCUGG...... KX510117.1.1407여기서 말하는 컬럼은 글자 하나. PDB파일처럼 얘도 알파벳 하나가 컬럼이다.
for record in alignment:
print(record.id, record.description) # 예제 코드는 Sequence와 ID를 출력하라고 했는데 기니까 다른걸로 바꿔보자.AB102732.157.1651 AB102732.157.1651 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Agrobacterium radiobacter
AB680818.1.1406 AB680818.1.1406 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Rhizobium tropici
AB680959.1.1462 AB680959.1.1462 Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Pseudomonadaceae;Pseudomonas;Agrobacterium agile
AB682466.1.1406 AB682466.1.1406 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Rhizobium hainanense
AB682468.1.1406 AB682468.1.1406 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Rhizobium gallicum
AB969785.1.1417 AB969785.1.1417 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Agrobacterium pusense
AJ786600.1.1449 AJ786600.1.1449 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Hoeflea;Hoeflea alexandrii
DQ835303.1.1353 DQ835303.1.1353 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Rhizobium fabae
GU564401.1.1419 GU564401.1.1419 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Hoeflea;Hoeflea halophila
JF957616.1.1410 JF957616.1.1410 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Hoeflea;Hoeflea phototrophica
JQ230000.1.1392 JQ230000.1.1392 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Ciceribacter;Ciceribacter lividus
KP142169.1.1341 KP142169.1.1341 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Agrobacterium salinitolerans
KU975391.1.1451 KU975391.1.1451 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Ciceribacter;Ciceribacter thiooxidans
KX510117.1.1407 KX510117.1.1407 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Ciceribacter azotifigens이런 식으로 레코드 만들어도 된다. 예제에는 ID랑 시퀀스로 되어 있던거 ID랑 description으로 바꿨다... 시퀀스가 너무 길어... 출력 format을 지정하거나 쌩으로 레코드를 가져올 수도 있는데, 길이가... 길이가...... 정말 장난없다...
from Bio import AlignIO
alignments = AlignIO.parse("/home/koreanraichu/lactobacillus.fasta", "fasta")
for alignment in alignments:
print(alignment)Alignment with 12 rows and 1606 columns
...............UUUGAUCAUGGCUCAGGACGAACGCUGGC...... AB008203.1.1553
................UUGAUCAUGGCUCAGGACGAACGCUGGC...... AB008210.1.1552
.........................................CGC...... AB022925.1.1450
..........................................GC...... AB023242.1.1446
............GAGUUUGAUCCUGGCUCAGGACGAACGCUGGC...... AB112083.1.1557
...........GGGUUUCGAUUCUGGCUCAGGAUGAACGCUGGC...... AB437354.1.1520
.............GUUUCGAUUCUGGCUCAGGAUGAACGCUGGC...... AB437356.1.1519
...............................GAUGAACGCUGGC...... AF173986.1.1505
...........AGAGUUUGAUCCUGGCUCAGGAUGAACGCUGGC...... AYZK01000017.137.1688
UUUUUUUGUGGAGGGUUUGAUUCUGGCUCAGGAUGAACGCUGGC...AGA CP011786.1420993.1422533
UUUUUUUGUGGAGGGUUUGAUUCUGGCUCAGGAUGAACGCUGGC...AGA CP011786.1514900.1516440
...............................GAUGAACGCUGGC...... KX232108.1.1480Parse로 가져와도 똑같긴 한데 SeqIO나 얘나 파싱으로 가져오면 for문 있어야 제대로 뜬다.
from Bio import AlignIO
handle="/home/koreanraichu/lactobacillus.fasta"
for alignments in AlignIO.parse(handle, "fasta",seq_count=2):
print("Alignment lentgh %i" % alignments.get_alignment_length())
for record in alignments:
print(record.description)Alignment lentgh 1606
AB008203.1.1553 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lactobacillus;Lactobacillus acidophilus
AB008210.1.1552 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lactobacillus;Lactobacillus helveticus
Alignment lentgh 1606
AB022925.1.1450 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Leuconostoc;Leuconostoc carnosum
AB023242.1.1446 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Leuconostoc;Leuconostoc mesenteroides
Alignment lentgh 1606
AB112083.1.1557 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lactiplantibacillus;Lactobacillus plantarum
AB437354.1.1520 Bacteria;Actinobacteriota;Actinobacteria;Bifidobacteriales;Bifidobacteriaceae;Bifidobacterium;Bifidobacterium adolescentis
Alignment lentgh 1606
AB437356.1.1519 Bacteria;Actinobacteriota;Actinobacteria;Bifidobacteriales;Bifidobacteriaceae;Bifidobacterium;Bifidobacterium bifidum
AF173986.1.1505 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Leuconostoc;Leuconostoc kimchii
Alignment lentgh 1606
AYZK01000017.137.1688 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lacticaseibacillus;Lactobacillus thailandensis DSM 22698 = JCM 13996
CP011786.1420993.1422533 Bacteria;Actinobacteriota;Actinobacteria;Bifidobacteriales;Bifidobacteriaceae;Bifidobacterium;Bifidobacterium actinocoloniiforme DSM 22766
Alignment lentgh 1606
CP011786.1514900.1516440 Bacteria;Actinobacteriota;Actinobacteria;Bifidobacteriales;Bifidobacteriaceae;Bifidobacterium;Bifidobacterium actinocoloniiforme DSM 22766
KX232108.1.1480 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Leuconostoc;Leuconostoc miyukkimchii끊어뽑기도 된다. 코드에 있는 숫자 수정하면 3개 4개 묶는 것도 된다.
쓰기
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio.Align import MultipleSeqAlignment
from Bio import AlignIO
align1=MultipleSeqAlignment([
SeqRecord(Seq("GGCC"),id="HaeIII"),
SeqRecord(Seq("G-CC"),id="id2"),
SeqRecord(Seq("G--C"),id="id3")
])
align2=MultipleSeqAlignment([
SeqRecord(Seq("GAATTC"),id="EcoRi"),
SeqRecord(Seq("G-ATTC"),id="id5"),
SeqRecord(Seq("G--TTC"),id="id6")
])
my_alignment=[align1,align2]
# 여기까지 시퀀스를 만들고
AlignIO.write(my_alignment, "/home/koreanraichu/sequence.fasta", "fasta")
# 여기서 저(별)장나 저거 IL-16 전에 썼던거 안된 이유 알아냈음... ㅋㅋㅋㅋㅋㅋ Seq=("")가 아니라 Seq("")였다. 아무튼 이런 식으로 쓸 수 있고 주석 있어서 대충 아시겠지만 순서대로 Alignment 요소를 만들고->시퀀스 만들고->파일로 저장하는 코드이다.
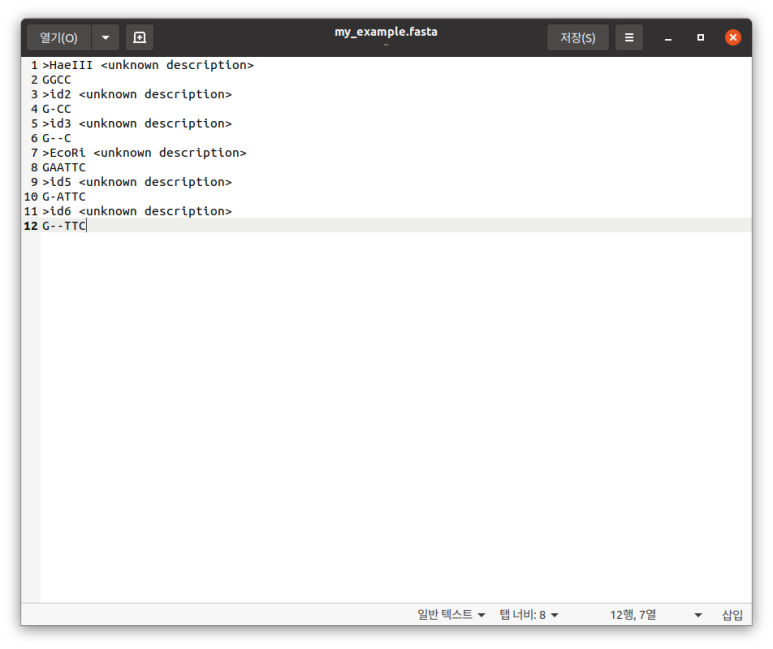
이런 식으로 저장된다. (그야 내가 FASTA로 저장했으니까...)
참고로 미리 말해두겠지만 본인 리눅스에서 FASTA, PHYLIP, 스톡홀름, clustalW 다 지에딧으로 연다. 그렇게 열리고…
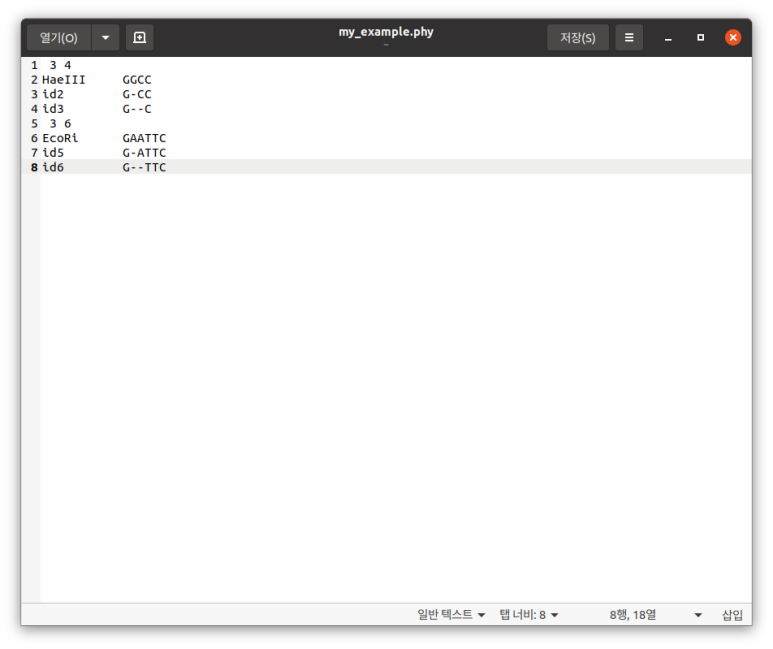
이게 필립 파일인데 얘는 변환하려면 시퀀스 길이가 다 같아야 하고, 공백이 -이어야 한다. 공백에 . 있으면 지원 안 한다고 오류 토한다.
선생님 이거 변환 됩니까?
변환이요? 뭐 클러스탈이나 스톡뭐시기 말하는거면 된다.
count = AlignIO.convert("/home/koreanraichu/agrobacterium.fasta", "fasta", "/home/koreanraichu/agrobacterium.sth",
"stockholm")
print("Converted %i alignments" % count)alignments = AlignIO.parse("/home/koreanraichu/agrobacterium.fasta", "fasta")
count = AlignIO.write(alignments, "/home/koreanraichu/agrobacterium.aln","clustal")
print("Converted %i alignments" % count)
# ClustalW위는 convert를 사용해 스톡홀름 파일로 바꾼거고, 아래는 parse와 write를 사용해 클러스탈로 바꾼 것이다.
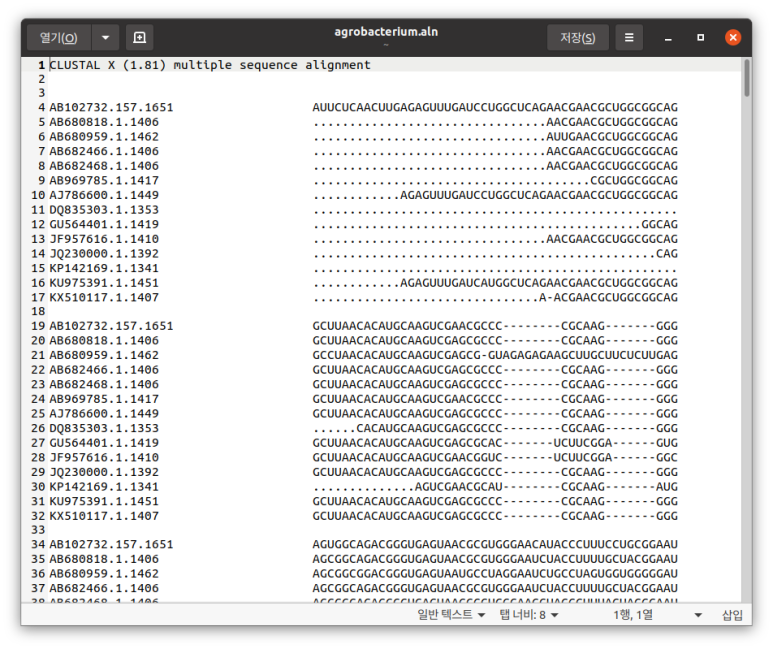
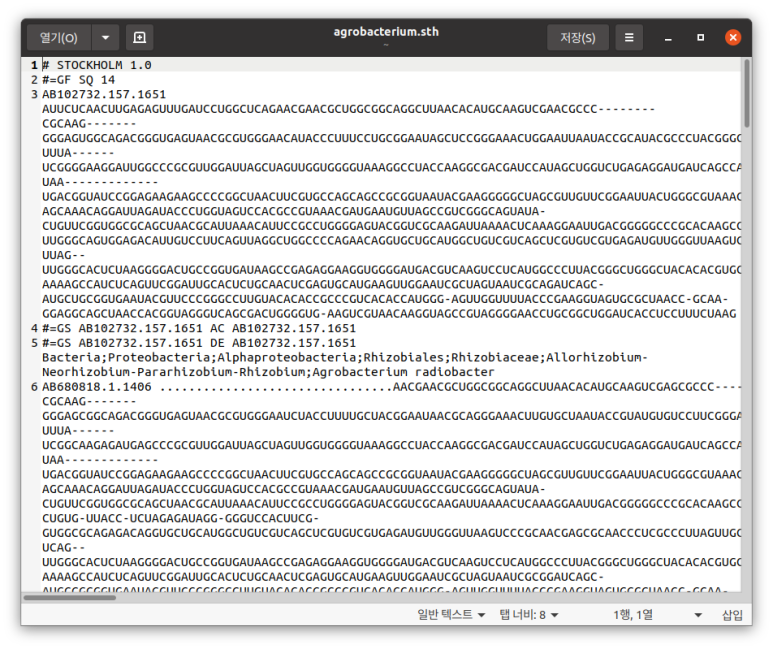
위가 clustalw, 아래가 스톡홀름 포맷.
alignment = AlignIO.read("/home/koreanraichu/PF00096_seed.txt", "stockholm")
AlignIO.write([alignment], "PF05371_seed.aln", "clustal")
print("Converted %i alignments" % count)
# read 후 리스트화해서 변환read로 읽은 다음 리스트화해서 변환하는 것도 된다.
count = AlignIO.convert("/home/koreanraichu/PF08449_seed.txt", "stockholm", "/home/koreanraichu/PF08449_seed.phy",
"phylip")
print("Converted %i alignments" % count)
# 이거라면 필립 될거같은데?공백이 -이고 시퀀스 bp가 전부 동일하다는 전제하에 Phylip 포맷으로 만드는 것도 된다.
alignment2 = AlignIO.read("/home/koreanraichu/PF08449_seed.txt", "stockholm")
name_mapping = {}
for i, record in enumerate(alignment):
name_mapping[i] = record.id
record.id = "seq%i" % i
AlignIO.write([alignment], "PF08449_seed_ID.phy", "phylip")
# 오 뭔진 모르겠지만 ID가 숫자가 된 건가딕셔너리화 한 다음 아이디 새로 지정해주는 것도 된다. 위 코드를 실행하면
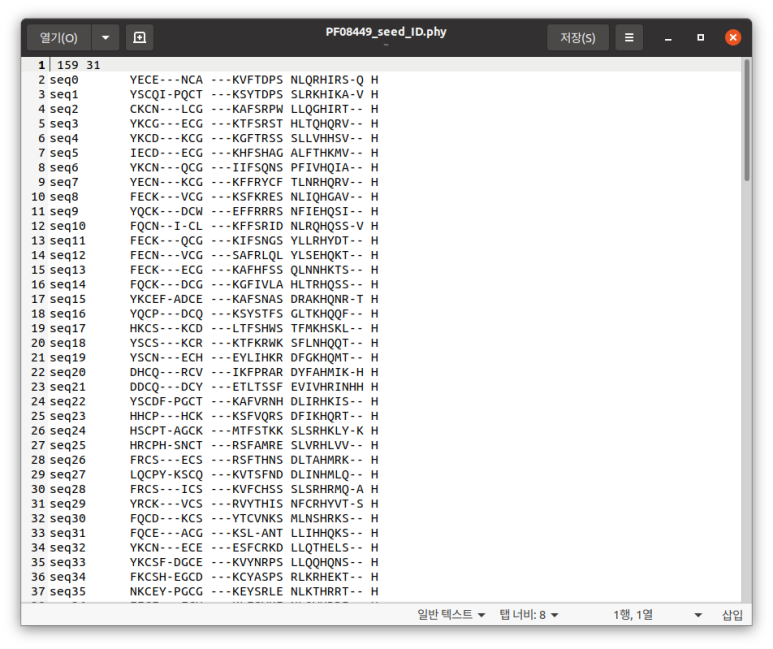
이렇게 된다. 잘 보면 ID 영역에 seq(숫자)가 들어가 있다.
출력 형식만 바꾸기
이건 뭔 소리냐... 여는 파일은 FASTA파일인데 파일 수정 없이 clustalw나 스톡홀름 형식으로 출력할 수 있다. (phylip은 아마 글자수 같고 공백 -여야 먹힐듯) 참고로 결과는 길어서 생략.
from Bio import AlignIO
alignment = AlignIO.read("/home/koreanraichu/lactobacillus.fasta", "fasta")
print(format(alignment, "clustal"))해당 코드는 FASTA파일을 클러스탈 형식으로 보여달라는 얘기고, 스톡홀름으로 볼 수도 있다.
슬라이싱
넘파이 배열 자르는것처럼 얘도 슬라이싱이 된다. (넘파이 2차원 배열 잘라먹는 거 생각하면 된다)
from Bio import AlignIO
alignment = AlignIO.read("/home/koreanraichu/PF08449_seed.txt", "stockholm")
print(alignment[0:5])ISLIPISMIMVGCCSNVISLELIMKQSQ-SH-A---------IL...TSG S35B4_DICDI/7-328
SFVLILSLVFGGCCSNVISFEHMVQGSN-INLG---------NI...YGS YEA4_KLULA/2-321
NSLKAFALVFGGCCSNVITFETLMSNET-GSIN---------NL...LGS YEA4_YEAST/3-327
MIASALSFIFGGCCSNAYALEALVREFP-SS-G---------IL...SAR YEA4_SCHPO/1-304
--GVMLSLIFGGCCSNVFALESIIKVEP-GA-G---------TL...L-- Q1K947_NEUCR/81-406이렇게 하면 pandas의 head()처럼 위에서 다섯개만 보여준다.
print(alignment[0,1]) # 0번째 인덱스의 두번째 컬럼
S이런 식으로 2차원 인덱싱과 슬라이싱도 되고
print(alignment[:,6]) # 무슨 컬럼을 가져온거냐
SSASSGTCLSSASLASL이거는 전체 인덱스에서 일곱번째 글자들을 가져온 것. (실화다)
print(alignment[0:5,0:5]) # index+column 범위를 지정할 수 있다
ISLIP S35B4_DICDI/7-328
SFVLI YEA4_KLULA/2-321
NSLKA YEA4_YEAST/3-327
MIASA YEA4_SCHPO/1-304
--GVM Q1K947_NEUCR/81-406이런 식으로 범위 지정해서 잘라오는 것도 되고 특정 인덱스, 컬럼부터 다 가져오는 것도 된다. 저거 저대로 잘라먹는 것도 되고, Numpy 데려와서 배열화하는 것도 가능하다. 근데 예제대로 했더니 오류남... (안되는 건 아닌데 오류뜬다)
본격적인 정보 얻기
아까까지 대체 뭐 한겨...
from Bio import AlignIO
alignment = AlignIO.read("/home/koreanraichu/lactobacillus.fasta", "fasta") # 아이고 유산균씨 오랜만입니다
substitutions = alignment.substitutions
print(substitutions) . A C G U
. 1365.0 321.0 310.5 461.5 406.0
A 321.0 19985.0 791.0 2218.5 978.5
C 310.5 791.0 18856.0 957.5 2048.0
G 461.5 2218.5 957.5 26642.0 1015.0
U 406.0 978.5 2048.0 1015.0 16007.0이거는 rRNA라 티민 대신 우라실이 나왔다. 저건 아마도 해당 염기가 다른걸로 바뀐 비율? 횟수? 이런 걸 봐주는 것 같다. 예를 들자면 아데닌이 시토신, 구아닌, 우라실로 바뀌는 뭐 그런거. (공백 공백은 뭐여...) 저기서 옵션으로 .select() 주면 출력 순서가 바뀐다. (예: .select(“AUGC.”)으로 주면 행렬 인덱스랑 컬럼 순서가 AUCG.로 바뀐다)
Tool
이 부분이 문제가 많아요... OTL 둘 다 깔고 해야 하나...
ClustalW
from Bio.Align.Applications import ClustalwCommandline
cline = ClustalwCommandline("clustalw2", infile="/home/koreanraichu/lactobacillus.fasta")
print(cline)이런 식으로 불러서
clustalw2 -infile=/home/koreanraichu/lactobacillus.fasta니가 왜 거기서 나옴???
이거 뭐 트리 만드는 파일 만들어준다매 왜 안해줘요. 이럴거면 그냥 메가 쓰면 안되냐
MUSCLE
from Bio.Align.Applications import MuscleCommandline
cline = MuscleCommandline(input="/home/koreanraichu/lactobacillus.aln", out="/home/koreanraichu/lactobacillus.txt")
print(cline)얘는 이런 식으로 부르면
muscle -in /home/koreanraichu/lactobacillus.aln -out /home/koreanraichu/lactobacillus.txt이러시는 이유가 있으실 것 아니예요.
from Bio.Align.Applications import MuscleCommandline
cline = MuscleCommandline(input="/home/koreanraichu/lactobacillus.fasta", out="/home/koreanraichu/lactobacillus.txt",
clw=True)
print(cline)
muscle -in /home/koreanraichu/lactobacillus.fasta -out /home/koreanraichu/lactobacillus.txt -clw이걸 쓰면 clustalW 비슷하게 출력된다. (물론 파일은 생기지 않았습니다)
from Bio.Align.Applications import MuscleCommandline
cline = MuscleCommandline(input="/home/koreanraichu/lactobacillus.fasta", out="/home/koreanraichu/lactobacillus.txt",
clwstrict=True)
print(cline)
muscle -in /home/koreanraichu/lactobacillus.fasta -out /home/koreanraichu/lactobacillus.txt -clwstrict이걸 쓰면 clustalW headline을 쓴다는데 파일이 없다. 착한 사람만 보이는 파일인가... 그래서 얘네는 못했다.
Pairwise
왜 블래스트에서 뭐 검색하면 결과에 줄 그어서 나오는 그거 말하는 거 맞다.
from Bio import pairwise2
from Bio import SeqIO
seq1 = SeqIO.read("/home/koreanraichu/alpha.faa", "fasta")
seq2 = SeqIO.read("/home/koreanraichu/beta.faa", "fasta")
alignments = pairwise2.align.globalxx(seq1.seq, seq2.seq)
print(alignments)근데 이거 파싱으로 개별 레코드끼리 비교 못하나... 나중에 다시 함 트라이해 볼 예정.
[Alignment(seqA='MV-LSPADKTNV---K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----T--PEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=217), Alignment(seqA='MV-LSPADKTNV--K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----T-PEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LSPADKTNV--K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----TP-EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LS-PADKTNV--K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-TP------EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=217), Alignment(seqA='MV-LSPADKTNV--K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHLTP------EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LSPADKTN-V-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----T-PEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LSPADKTNV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----TPEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=215), Alignment(seqA='MV-LS-PADKTNV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-TP-----EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LSPADKTNV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHLTP-----EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=215), Alignment(seqA='MV-LSPADKT-NV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----TPE-EKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LS-PADKTNV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-뭔진 모르겠지만 제가 잘못했습니다. (저거 len으로 보면 길이 장난없다)
print(pairwise2.format_alignment(*alignments[0]))이렇게 쳐 주면 비로소 우리가 생각하는(?)
MV-LSPADKTNV---K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-
|| | | | | | |||| | | ||| | | | | | | | | | | | | ||||| | | | || | | | | || | | || ||| || | | || | || |||| | | | | | | ||
MVHL-----T--PEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H
Score=72이 결과가 나온다. BLAST에서도 이런 식으로 나온다.
BLOSUM62와 함께 정렬...아니고 줄을
from Bio import pairwise2
from Bio import SeqIO
from Bio.Align import substitution_matrices
blosum62 = substitution_matrices.load("BLOSUM62")
seq1 = SeqIO.read("/home/koreanraichu/alpha.faa", "fasta")
seq2 = SeqIO.read("/home/koreanraichu/beta.faa", "fasta")
alignments = pairwise2.align.globalds(seq1.seq, seq2.seq,blosum62, -10, -0.5)
print(pairwise2.format_alignment(*alignments[0]))이거 [0] 빼면요? 뭔진 모르겠지만 잘못했어요 소리가 절로 나온다. (길이 장난없음)
MV-LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS-----HGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR
|| |.|..|..|.|.|||| ...|.|.|||.|.....|.|...|..| ||| .|...||.|||||..|.....||.|........||.||..||.|||.||.||...|...||.|...||||.|.|...|..|.|...|..||.
MVHLTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH
Score=292.5.globalds는 전역(전체 시퀀스)를 보는거고 .localds로 부분만 볼 수도 있다.
alignments = pairwise2.align.localds("LSPADKTNVKAA", "PEEKSAV", blosum62, -10, -1)3 PADKTNV
|..|..|
1 PEEKSAV
Score=16이런 식으로 부분부분 볼 수도 있다. 엥? 근데 저렇게 해 놓으면 어딘지 어떻게 알아요?
print(pairwise2.format_alignment(*alignments[0],full_sequences=True))LSPADKTNVKAA
|..|..|
--PEEKSAV---개발자는 다 계획이 있었다.
Pairwise aligner
from Bio import Align
aligner = Align.PairwiseAligner()
seq1="GAATTC"
seq2="GCATTC"
score = aligner.score(seq1, seq2)
print(score)
5.0일단 모셔보래서 모셔봤습니다. 이거는 score 뽑는거고
from Bio import Align
aligner = Align.PairwiseAligner() # 그래서 모셔왔습니다
seq1="GAATTC"
seq2="GCATTC"
alignments = aligner.align(seq1, seq2)
for alignment in alignments:
print(alignment)이렇게 치면
G-AATTC
|-|-|||
GCA-TTC
GA-ATTC
|--||||
G-CATTC
G-AATTC
|--||||
GC-ATTC
GAATTC
|.||||
GCATTC이렇게 나온다. 전 맨 밑에껄로 보여주세요. 픽 안되나
GAATTC
||||||
GAATTC100% 일치하는 시퀀스는 이런 식으로 보여준다. aligner.mode="local"을 주면 위에것처럼 중구난방으로 안 뽑아주고
GAATTC
|||||
CAATTC이런 식으로 뽑아준다.
Wildcard
일반적으로 검색할 때(구글이나 파일같은 거) *이 와일드카드지만, 여기서는 와일드카드를 지정할 수 있다.
# wildcard를 지정한 다음 쓸 수도 있다
aligner.wildcard = "?"
seq3="?GCC"
seq4="GGCC"
alignments2 = aligner.align(seq3, seq4)
for alignment in alignments2:
print(alignment)?G-CC
|-||
GGCC
?GCC
|||
GGCC여기서는 일단 ?로 지정해보자.
'Coding > Python' 카테고리의 다른 글
| Biopython으로 BLAST 돌려보기 (0) | 2022.08.20 |
|---|---|
| 번외편-코딩테스트 풀이 (0) | 2022.08.20 |
| Biopython SeqIO 써보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 레코드 생성하고 만져보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 다뤄보기 (0) | 2022.08.20 |