풀긴 풀었는데 이거 로직이 뭐가 문제인건지 제시한거랑 출력값이 다름.
일단 문제가 뭐냐... 예를 들어서 아래와 같은 텍스트가 있다면
츄라이츄라이출력값은
000123이 된다. 네 번째 글자 츄를 기준으로 했을 때
츄라이에서 츄/이츄/라이츄를 찾는건데 저기서 일치하는 게 츄 하나고 그게 한글자짜리거든.
다섯번째 글자 라를 기준으로 하면 츄라이츄에서 라/츄라/이츄라/라이츄라 이렇게 찾게 되는거고 그렇게 되면 일치하는 문자열 중 제일 긴 게 츄라라서 2.
이해하는 데 하루 걸렸다더니 실화냐고요? 네. 잠깐 뇌에 블루스크린 버프 와서요.
text=input("Insert text: ")
# 전체 텍스트를 slicing하는 for문. 이 안에는 찾을 영역과 찾아야 할 영역이 포함되어 있다.
# 한 글자일때는 찾을 영역이 존재하지만 찾아야 하는 글자가 없으므로 첫 글자는 0이다.
find_list=[]
length=len(text)
for i in range(1,length+1):
if len(text[0:i]) == 1:
find_list.append(0)
else:
text_sub=text[0:i] # 전체 텍스트
find=text_sub[0:i-1]# 찾을 범위
max_list=[]
for j in range(1,len(find)+1):
subset=text_sub[j:] # 찾을 텍스트
find_values=0
if subset in find:
max_list.append(len(subset))
else:
max_list.append(0)
max_list=set(max_list)
max_list=list(max_list)
max_values=max(max_list) # 리스트에서 최대값을 추출
find_list.append(max_values) # 했으면 넣어주세요
# 리스트 출력이 뭔가 이상하다.
# set을 적용하게 되면 각 iteration별로 고유값이 나오는 게 아니라 엉뚱한 값이 나오게 된다.
# append의 대상이 되는 리스트는 밖에 있지만, append는 안에 있어서 또 애매하고... append를 밖으로 뺄 수도 없다.
find_text=''.join(map(str,find_list))
# 리스트 형태로 처리한 것을 문자열로 붙여서 출력
print(find_text)일단 전체 코드는 이거.
사실 문제가 정말 궁서체로 개빡세다. 어제 추워서 쉬긴 했지만 그래도 이틀 걸렸다. (주말에는 공부 안 함) 코딩은 컴퓨터 시켜먹으려면 일단 사람이 이해를 해야 하는데 이해하고 착수하는데 꽤 걸림+이게 내 맘대로 안돼서 걸림...
위에 설명한 게 문제인데, 그럼 필요한 기능이 뭐냐...
1. 리스트의 인덱싱/슬라이싱(각각 필요한 부분을 단식/범위로 찾는 기능)
2. For(반복문)
3. if(제어문)
4. 특정 문자를 찾는 기능(대충 R의 grep같은 거)
5. 입력(이건 맨 나중이라 우선순위 미뤄도 된다)
크게 이렇게 필요하다.
그럼 차근차근 게임코딩빨로 해봅시다. 이사람 대체 근데 솔직히 두뇌풀가동 이후로 엑스트라 밀고 겜코딩 켠 적 없음
Indexing/slicing 일반화 및 찾을 영역 정의하기
일단 본인은 별찍기때도 그러했듯 small scale(고정값 혹은 실행하는 데 오래 안 걸리는 작은 범위)에서 해보고 일반화 과정을 거친다. 대충 수열로 치자면 어떤 규칙으로 변하는지 몇 개 찍어보고 일반항 공식 도출하는 방식이다. (별찍기 그렇게 한 다음 while로 찍었더니 while이 더 편하더라...)
어떤 n글자짜리 문자열이 있을 때
-이 문자열의 인덱싱 주소는 0부터 (n-1)이고
-이 문자열의 슬라이싱 주소는 0부터 n까지이다. 이게 왜 그러냐... 아래 텍스트를 예시로 들어보자.
>>> text="HAPPY DAYS!"
>>> len(text)
11
>>> print(text[0:11])
HAPPY DAYS!H,A,P,P,Y, ,D,A,Y,S,!, 해서 11자(중간에 오타가 아니라 공백이 두 개 이고 이 때 인덱싱 주소는 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10이다. 근데 왜 11까지냐... 0에서 10까지니까 슬라이싱을 0:10으로 해 보면
>>> print(text[0:10])
HAPPY DAYS뭐야 느낌표 돌려줘요!!!
이게 왜 이렇게 되는거냐면 파이썬 슬라이싱은 [부터:미만]으로 들어가기 때문이다. 그래서 0:10으로 하면 0부터 10 미만, 즉 9까지 뽑아준다.
# 전체 텍스트를 slicing하는 for문. 이 안에는 찾을 영역과 찾아야 할 영역이 포함되어 있다.
# 한 글자일때는 찾을 영역이 존재하지만 찾아야 하는 글자가 없으므로 첫 글자는 0이다.
for i in range(1,length+1):
if len(text[0:i]) == 1:
print(0)
else:
text_sub=text[0:i] # 전체 텍스트
find=text_sub[0:i-1]# 찾을 범위
print(find) # 이게 찾을 범위이게 찾는 범위를 지정하는 코드. 왜 1부터냐... 저게 [부터:미만]이라고 했는데 일단 첫 빠따는 뭐가 들어가던 0이다. 왜냐, 찾을 영역은 지정할 수 있는데 찾을 글자가 없다. 뭔 소리냐...
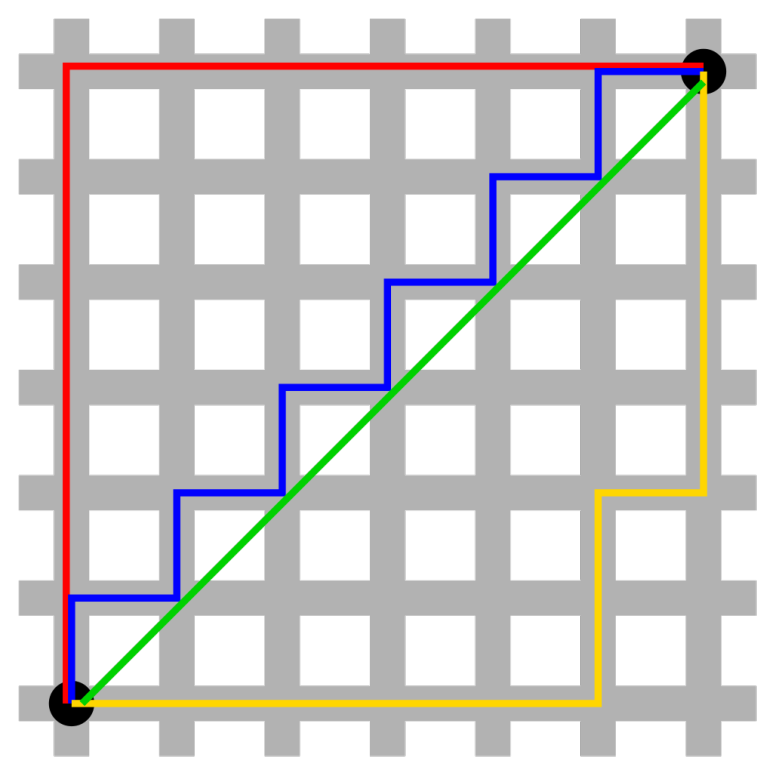
[0,1,2,3,4,5]위와 같은 리스트가 있을 때, 찾을 범위는 [0], [0,1],...,[0,4]까지이고 찾아야 할 글자는 범바범인데 범위가 [0]일 때 [1], [0,1]일 때 [2],[1,2] 이런 식으로 들어간다. 즉 찾는 영역은 앞에서부터 순차적으로 들어가고 찾아야 하는 영역은 뒤에서부터 순차적으로 들어간다.
일차적인 for문에서 range가 1,len(text)+1인 이유도 그것때문이다. 저거 없이 그냥 박아버리면 0부터 시작하기때문에 나중에 음수 인덱싱을 하게 될 경우 문제가 생긴다. (-0이나 0이나 그게 그거라...) 그리고 length+1 해야 슬라이싱하면 length값까지 잡아준다.
찾아야 할 부분집합 슬라이싱하기
# 전체 텍스트를 slicing하는 for문. 이 안에는 찾을 영역과 찾아야 할 영역이 포함되어 있다.
# 한 글자일때는 찾을 영역이 존재하지만 찾아야 하는 글자가 없으므로 첫 글자는 0이다.
for i in range(1,length+1):
if len(text[0:i]) == 1:
print(0)
else:
text_sub=text[0:i] # 전체 텍스트
find=text_sub[0:i-1]# 찾을 범위
subset_init=text_sub[i-1]
print(subset_init) # 첫 서브셋이자 시발점이 부분이 1차 난관이었다. 찾아야 하는 부분집합의 개수는 찾아야 할 영역의 크기에 따라 순차적으로 줄어들고, 찾아야 할 영역의 크기는 문자열의 길이에 따라 달라진다. 위에서 깜빡하고 안 썼는데, 이것은 마치 김밥 한 줄을 썰어서 왼쪽 꼬다리에서부터 오른쪽 꼬다리를 찾는 것과 비슷한 이치. 그러면 범위는 일단 김밥 한 줄로 잡아야 한다.
이 때 전체 텍스트는 김밥 한 줄, 찾을 부분은 왼쪽 꼬다리라고 보면 된다. 찾는 부분집합은 찾는 영역에 따라 다르지만 김밥 한 줄 기준으로 오른쪽 꼬다리부터 하나씩 시작이다. (찾을 범위에서 오른쪽 꼬다리가 빠지고, 부분집합을 만드는 범위에서 왼쪽 꼬다리가 빠진다)
그러니까 김밥 한 줄에서 오른쪽 꼬다리부터 순차적으로 지정해야 해서 원래 저 안에 for문이 또 들어간다. 그래서 전체 코드를 보면 for문이 두 개 들어간 것. ...저 시발점을 잘못 잡은 게 오른쪽 꼬다리면 무조건 -1번 인덱싱(맨 뒤)인데...
제어문
# 전체 텍스트를 slicing하는 for문. 이 안에는 찾을 영역과 찾아야 할 영역이 포함되어 있다.
# 한 글자일때는 찾을 영역이 존재하지만 찾아야 하는 글자가 없으므로 첫 글자는 0이다.
find_subset=[]
max_subset=[]
for i in range(1,length+1):
if len(text[0:i]) == 1:
find_subset.append(0)
print(0)
else:
text_sub=text[0:i] # 전체 텍스트
find=text_sub[0:i-1]# 찾을 범위
for j in range(1,len(find)+1):
subset=text_sub[1:j+1] # 찾을 텍스트
# 텍스트 유무에 따른 처리는 했는데, 문제는 텍스트가 있을 때 길이가 아니라 리스트 자체가 출력된다... 이거 어쩔겨...
if subset in find:
print(subset)
find_subset.append(1)
else:
find_subset.append(0)
print(find_subset)[0, 0, 1, 0, 1, 1, 0, 1, 1, 1, 0, 1, 1, 1, 1, 0, 1, 1, 1, 1, 1, 0, 1, 1, 1, 1, 1, 1, 0](마른세수)
출력이 개판인데 그거는 이따가 또 얘기합시다... 진짜 출력 잡는것도 대환장파티였어요...
아무튼 저기 안쪽에 for문 하나 더 들어간 게 찾아야 할 부분집합이라는 건 알겠는데, if가 왜 있느냐... 일단 저 코드는 일치하는 게 있으면 1, 없으면 0을 끼워 넣으라는 의미긴 하다. 그러니까 쉽게 말하자면 일치하는 게 있냐 없냐를 먼저 보면서 범위부터 손볼 요량이었다.
if subset in find:
print(subset)
max_subset.append(1)
find_subset.append(len(max_subset))
else:
find_subset.append(0)제어문은 일단 이런 식. 해당하는 텍스트가 있을 때 일치하는 텍스트의 '길이'를 넣는다.
if subset in find:
max_list.append(len(subset))
else:
max_list.append(0)
max_list=set(max_list)
max_list=list(max_list)
max_values=max(max_list) # 리스트에서 최대값을 추출전체 코드에서는 이런 식으로 처리한다.
출력 처리
여기서 정말 마른세수 여러번 했다... (마른세수)
# 전체 텍스트를 slicing하는 for문. 이 안에는 찾을 영역과 찾아야 할 영역이 포함되어 있다.
# 한 글자일때는 찾을 영역이 존재하지만 찾아야 하는 글자가 없으므로 첫 글자는 0이다.
find_subset=[] # 서브셋들의 최대값을 담아서 최종적으로 출력하는 리스트
max_subset=[] # 서브셋들의 최대값을 매기기 위한 리스트
for i in range(1,length+1):
if len(text[0:i]) == 1:
find_subset.append(0)
else:
text_sub=text[0:i] # 전체 텍스트
find=text_sub[0:i-1]# 찾을 범위
for j in range(1,len(find)):
print(j,"th iteration")
subset=text_sub[j:] # 찾을 텍스트
print(find,len(find))
print(subset,"*")
if subset in find:
max_subset.append(len(subset))
max_values=max(max_subset)
print(max_subset,max_values)
find_subset.append(max(max_subset))
print(find_subset)
else:
find_subset.append(0)
print(find_subset)
# 리스트 출력이 뭔가 이상하다. 이거 중복값 처리를 어떻게 해야 하는거지?이놈은 돌려봤더니 중복값이 고대로 나오고...
# 전체 텍스트를 slicing하는 for문. 이 안에는 찾을 영역과 찾아야 할 영역이 포함되어 있다.
# 한 글자일때는 찾을 영역이 존재하지만 찾아야 하는 글자가 없으므로 첫 글자는 0이다.
find_list=[]
for i in range(1,length+1):
if len(text[0:i]) == 1:
find_list.append(0)
else:
text_sub=text[0:i] # 전체 텍스트
find=text_sub[0:i-1]# 찾을 범위
for j in range(1,len(find)):
subset=text_sub[j:] # 찾을 텍스트
find_values=0
if subset in find:
find_list.append(len(subset))
else:
find_list.append(0)
find_list=set(find_list)
find_list=list(find_list)
# 리스트 출력이 뭔가 이상하다.
print(find_list)이놈은 돌렸더니 중복값이 다 사라져서 글자수랑 안 맞고...
일단 iteration의 기준이 뭐냐면, 찾을 범위를 하나 지정하고 거기서 문자열 차즌ㄴ 게 하나의 iteration이다. 즉, 첫번째 for문을 돌 때마다 값이 하나씩 저장되어야 한다.
# 전체 텍스트를 slicing하는 for문. 이 안에는 찾을 영역과 찾아야 할 영역이 포함되어 있다.
# 한 글자일때는 찾을 영역이 존재하지만 찾아야 하는 글자가 없으므로 첫 글자는 0이다.
find_list=[]
for i in range(1,length+1):
if len(text[0:i]) == 1:
find_list.append(0)
else:
text_sub=text[0:i] # 전체 텍스트
find=text_sub[0:i-1]# 찾을 범위
max_list=[]
for j in range(1,len(find)+1):
subset=text_sub[j:] # 찾을 텍스트
find_values=0
if subset in find:
max_list.append(len(subset))
else:
max_list.append(0)
max_list=set(max_list)
max_list=list(max_list)
max_values=max(max_list)
find_list.append(max_values)
# 리스트 출력이 뭔가 이상하다.
# set을 적용하게 되면 각 iteration별로 고유값이 나오는 게 아니라 엉뚱한 값이 나오게 된다.
# append의 대상이 되는 리스트는 밖에 있지만, append는 안에 있어서 또 애매하고... append를 밖으로 뺄 수도 없다.
find_text=''.join(map(str,find_list))
print(find_text)근데 이게 이거 뺀다고 해결되는거 실화냐... 아, 저기 join은 출력을 리스트 형태로 하기 위해 넣은거다.
AGTC AGTC CGTG ATCT AGCT AGCT AGTC GCTG CATC AGTA C
0000 1234 1121 1121 1212 3456 7834 2232 2223 3452 1 # 돌려서 나온 결과
0000 1234 0000 1000 1200 1200 1234 0000 0100 1231 0 # 문제에서 제시한 결과근데 이거 왜 결과가 이렇게 나오지...?
'Coding > Python' 카테고리의 다른 글
| 오케이 따옴표 떼버렸음 (0) | 2022.08.21 |
|---|---|
| 10진수->2진수 변환 코드 (0) | 2022.08.21 |
| 번외편-코딩테스트 풀이 (2) (0) | 2022.08.21 |
| Biopython-dbSNP와 Clinvar (0) | 2022.08.21 |
| 심심해서 써보는 본인 개발환경 (0) | 2022.08.21 |