

- R 배워보기- 6.5. Manipulating data-Sequential data 2022.08.20
- R 배워보기- 6.4. Manipulating data-Restructing data 2022.08.20
- R 배워보기- 6.3. Manipulating data-Data Frames 2022.08.20
- R 배워보기- 6.2. Manipulating data-Factors 2022.08.20
- R 배워보기- 6.1. Manipulating data-General 2022.08.20
- Biopython으로 MSA 해보기 2022.08.20
- Biopython SeqIO 써보기 2022.08.20
- Biopython으로 시퀀스 레코드 생성하고 만져보기 2022.08.20
- Biopython으로 시퀀스 다뤄보기 2022.08.20
- Biopython으로 시퀀스 가져오기 2022.08.20
내일 예고: 통계분석 들어가기 때문에 골치아파질 예정 교수님 죄송합니다 여러번 외칠 예정
이동평균 계산하기
이동평균: 전체 데이터 집합의 여러 하위 집합에 대한 일련의 평균을 만들어 데이터 요소를 분석하는 계산(솔직히 뭐 하는건지는 모르겠음)
난 sequential data라길래 파이썬처럼 시퀀스형 데이터가 있나 했더니 연속형 데이터 말하는건가봄. 전구간에서 미분 가능한가요 NA 들어가면 짤없을 예정
> set.seed(1)
> x=1:300
> y=sin(x)+rnorm(300,sd=1)
> y[295:300]=NA
> plot(x, y, type="l", col=grey(.5))일단 뒤에 여백의 미를 줄 예정이다.

(마른세수)
> grid()
이게 모눈을 킨다고 다 이쁜 그래프가 아니그등요... 아무튼 계산해보자...
> f20=rep(1/20,20)
> f20
[1] 0.05 0.05 0.05 0.05 0.05 0.05 0.05 0.05 0.05 0.05 0.05 0.05 0.05 0.05 0.05
[16] 0.05 0.05 0.05 0.05 0.05
> y_lag=filter(y,f20,sides=1)
> lines(x,y_lag,col="red")
> f21=rep(1/21,21)
> f21
[1] 0.04761905 0.04761905 0.04761905 0.04761905 0.04761905 0.04761905
[7] 0.04761905 0.04761905 0.04761905 0.04761905 0.04761905 0.04761905
[13] 0.04761905 0.04761905 0.04761905 0.04761905 0.04761905 0.04761905
[19] 0.04761905 0.04761905 0.04761905
> y_sym=filter(y,f21,sides=2)
> lines(x,y_sym,col="blue")
솔직히 왜 20, 21인지는 모르겠다. 쿡북에 sin(x/20)으로 되어 있어서 그런가...
아 참고로

filter()는 결측값이 있으면 공백이 된다. 여백의 미
블록으로 나눠서 평균 매기기
> set.seed(3)
> x=floor(runif(22)*100)
> x
[1] 16 80 38 32 60 60 12 29 57 63 51 50 53 55 86 82 11 70 89 27 22 1runif()=난수 생성, floor()=소수점 떼뿌라!!!
# Round up the length of vector the to the nearest 4
> newlength=ceiling(length(x)/4)*4
> newlength
[1] 24위에서 만든 벡터에서 네개씩 묶어서 평균 낸 결과라고 한다. (솔직히 귀찮아서 검산 안했음)
> x[newlength]=NA
> x
[1] 16 80 38 32 60 60 12 29 57 63 51 50 53 55 86 82 11 70 89 27 22 1 NA NA일단 길이를 늘리기 위해 벡터 두 개를 끼워넣고
> x=matrix(x,nrow=4)
> x
[,1] [,2] [,3] [,4] [,5] [,6]
[1,] 16 60 57 53 11 22
[2,] 80 60 63 55 70 1
[3,] 38 12 51 86 89 NA
[4,] 32 29 50 82 27 NA이것이 행렬이다 희망편(아님)... 은 아니고 matrix()로 행렬 만들었다. 그러면
> colMeans(x,na.rm=TRUE)
[1] 41.50 40.25 55.25 69.00 49.25 11.50
# 컬럼 평균
> rowMeans(x,na.rm=TRUE)
[1] 36.50000 54.83333 55.20000 44.00000
# 열 평균아까랑 다른데?
rle()
rle가 아마 run length encoding의 약자인듯. 자꾸 나와 이거...
> v <- c("A","A","A", "B","B","B","B", NA,NA, "C","C", "B", "C","C","C")
> v
[1] "A" "A" "A" "B" "B" "B" "B" NA NA "C" "C" "B" "C" "C" "C"여기 벡터가 있다.
> vr=rle(v)
> vr
Run Length Encoding
lengths: int [1:7] 3 4 1 1 2 1 3
values : chr [1:7] "A" "B" NA NA "C" "B" "C"rle()를 줘 보면 NA가 각개로 나오는 것을 볼 수 있다. 얘는 python의 set과 달리 끊어져 있으면 중복으로 안 치는 듯 하다. (python set은 중복이면 가차없이 빼버림)
> inverse.rle(vr)
[1] "A" "A" "A" "B" "B" "B" "B" NA NA "C" "C" "B" "C" "C" "C"inverse.rle()를 쓰면 다시 벡터로 돌릴 수 있다.
> str(w)
chr [1:11] "Alpha" "Alpha" "pi" "pi" "pi" "Omicron" "Psi" "Psi" "Xi" "Xi" ...
> str(rle(w))
List of 2
$ lengths: int [1:5] 2 3 1 2 3
$ values : chr [1:5] "Alpha" "pi" "Omicron" "Psi" ...
- attr(*, "class")= chr "rle"확인해보면 타입이 다르다. (rle는 리스트 두 개)
> x=v
> x[is.na(x)]="D"
> x
[1] "A" "A" "A" "B" "B" "B" "B" "D" "D" "C" "C" "B" "C" "C" "C"NA를 다른 걸로 대체하게 되면
> xr=rle(x)
> xr
Run Length Encoding
lengths: int [1:6] 3 4 2 2 1 3
values : chr [1:6] "A" "B" "D" "C" "B" "C"rle() 결과가 달라진다. NA가 다 D로 바뀌었기 때문.
> xr$values[xr$values=="D"]=NA
> xr
Run Length Encoding
lengths: int [1:6] 3 4 2 2 1 3
values : chr [1:6] "A" "B" NA "C" "B" "C"그 상태에서 D를 NA로 바꾼 것은 원래 벡터를 그냥 rle() 한 것과 또 다르다.
> x2=inverse.rle(xr)
> x2
[1] "A" "A" "A" "B" "B" "B" "B" NA NA "C" "C" "B" "C" "C" "C"
# 중간에 NA를 때웠던 벡터
> inverse.rle(vr)
[1] "A" "A" "A" "B" "B" "B" "B" NA NA "C" "C" "B" "C" "C" "C"
# NA 안 때운 벡터근데 벡터되면 다 똑같음.
이거 팩터에서도 되긴 되는데...
> f=factor(v)
> f
[1] A A A B B B B <NA> <NA> C C B C C C
Levels: A B C여기 팩터가 있다.
> f_levels=levels(f)
> f_levels
[1] "A" "B" "C"팩터의 레벨에 결측값은 낄 수 없다. ???: 뭐어? 결측값? 결측값??? 결측값도 레벨에 끼면 소는 누가 키울거야 소는! 결측값이랑 소랑 뭔 상관이여
> fc=as.character(f)
> fc
[1] "A" "A" "A" "B" "B" "B" "B" NA NA "C" "C" "B" "C" "C" "C"아무튼 얘는 글자로 변환하고 rle()를 준다. 사실상 이 뒤로는 벡터랑 같은 부분이라 생략.
근데 팩터에 rle 주면 안되냐고?
> rle(f)
Error in rle(f) : 'x'는 반드시 atomic 타입으로 이루어진 벡터이어야 합니다오류남.
바로 앞 데이터로 NA 채우기
> x <- c(NA,NA, "A","A", "B","B","B", NA,NA, "C", NA,NA,NA, "A","A","B", NA,NA)
> x
[1] NA NA "A" "A" "B" "B" "B" NA NA "C" NA NA NA "A" "A" "B" NA NA여기 벡터가 있다. (마른세수)
> goodIdx=is.na(x)
> goodIdx
[1] TRUE TRUE FALSE FALSE FALSE FALSE FALSE TRUE TRUE FALSE TRUE TRUE
[13] TRUE FALSE FALSE FALSE TRUE TRUE와씨 더럽게 많네...
> goodVals <- c(NA, x[goodIdx])
> goodVals
[1] NA "A" "A" "B" "B" "B" "C" "A" "A" "B"
# These are the non-NA values from x only
# Add a leading NA for later use when we index into this vectorNA 직전 값들을 알아낸다. (NA 앞 인덱스이면서 is.na()가 false인 값)
> fillIdx=cumsum(goodIdx)+1
> fillIdx
[1] 1 1 2 3 4 5 6 6 6 7 7 7 7 8 9 10 10 10
# 1을 더하는 이유는 0부터 채우는 걸 피하기 위함이다!그리고 채우기 위한 준비를 해 봅니다.
> goodVals[fillIdx]
[1] NA NA "A" "A" "B" "B" "B" "B" "B" "C" "C" "C" "C" "A" "A" "B" "B" "B"
# The original vector with gaps filled는 맨 앞에 두 개 빼고 때웠음.
'Coding > R' 카테고리의 다른 글
| R 배워보기-7. Statistical analysis (하) (0) | 2022.08.21 |
|---|---|
| R 배워보기-7. Statistical analysis (상) (0) | 2022.08.21 |
| R 배워보기- 6.4. Manipulating data-Restructing data (0) | 2022.08.20 |
| R 배워보기- 6.3. Manipulating data-Data Frames (0) | 2022.08.20 |
| R 배워보기- 6.2. Manipulating data-Factors (0) | 2022.08.20 |
들어가기 전에
아니 새기들아 깔아야 하는 라이브러리가 있으면 미리 좀 알려달라고!!! (깊은 분노)
아니 어느 레시피에서 재료설명도 없이 주저리 주저리 레시피 쓰다가 존내 당연하다는 듯 여러분 다들 집에 맨드레이크 있으시죠? 맨드레이크를 채썰어주세요. 하면서 레시피를 쓰냐!!! 집에 왜 그런게 있죠 아니 외가에서 무 받아온게 사람 모양이더라고
아무튼... 좀 개빡치긴 했지만... 라이브러리 깔고 가세요...
install.packages("tidyr")
install.packages("reshape2")
install.packages("doBy")테이블 가로세로 바꾸기
테이블은 보통 가로로 길거나 세로로 길거나 둘 중 하나이다. 캡처는 못했지만, 전전직장에서 일하면서 SQL로 정리해뒀던 샘플 표는 가로로 정말 엄청나게 긴 표였다. (시료의 색깔, 크기, 규격, 회사명, 제품명, 합불여부까지 다 기재해서... 마스크는 똑같은 회사에서 만들더라도 KF 규격이 다르거나 색, 크기가 다르면 다 검사 받아야 한다)
아무튼... 이게 가로세로가 바뀐다고?
> df=read.csv('/home/koreanraichu/example.csv',sep=";")
> df
ID Interesred.in Class
1 kimlab0213 Python Basic
2 ahn_0526 Python Medium
3 peponi01 R Basic
4 kuda_koma R Expert
5 comma_life Java Basic
6 wheresjohn Java Medium
7 hanguk_joa Python Expert
8 sigma_00 R Basic
9 kokoatalk Java Basic이건 근데 가로로 긴거냐 세로로 긴거냐... 그냥 세로로 방대한거 아닌가
> df_long=gather(df,ID,Interested.in,Class,factor_key=TRUE)
> df_long
ID Interested.in
1 Class Basic
2 Class Medium
3 Class Basic
4 Class Expert
5 Class Basic
6 Class Medium
7 Class Expert
8 Class Basic
9 Class Basictidyr 라이브러리의 gather()를 이용하면 이렇게 된다. (코드에는 생략되어 있으나 이 글을 읽는 여러분들이라면 아시리라 믿는다. tidyr 먼저 부르자)
> library(reshape2)
다음의 패키지를 부착합니다: ‘reshape2’
The following object is masked from ‘package:tidyr’:
smiths
> df_melt=melt(df,id.vars=c("ID","Class"))
> df_melt
ID Class variable value
1 kimlab0213 Basic Interested.in Python
2 ahn_0526 Medium Interested.in Python
3 peponi01 Basic Interested.in R
4 kuda_koma Expert Interested.in R
5 comma_life Basic Interested.in Java
6 wheresjohn Medium Interested.in Java
7 hanguk_joa Expert Interested.in Python
8 sigma_00 Basic Interested.in R
9 kokoatalk Basic Interested.in Javareshape2의 melt()로는 이렇게 한다.
> df_melt=melt(df,id.bvars=c("Interested.in","Class"),measure.vars=c("Interested.in","Class"),variable.names="Lenguages",value.name="ID")디테일한 지정도 된다.
이거는 넓은 표를 길게 했다 칩시다... 그럼 긴 표를 넓게 하려면 어떻게 해야 할까?
> df_wide=spread(df,ID,Class)
> df_wide
Interested.in ahn_0526 comma_life hanguk_joa kimlab0213 kokoatalk kuda_koma
1 Java <NA> Basic <NA> <NA> Basic <NA>
2 Python Medium <NA> Expert Basic <NA> <NA>
3 R <NA> <NA> <NA> <NA> <NA> Expert
peponi01 sigma_00 wheresjohn
1 <NA> <NA> Medium
2 <NA> <NA> <NA>
3 Basic Basic <NA>> df2_wide=spread(df2,Category.1,Price)
> df2_wide
ID Product.name Category.2 Order Cancel Consumable
1 BC-001 BCIG agar Bacteria 10 2 NA
2 DM-001 DMEM Cell culture 20 0 NA
3 GL-001 Slide glass Experimental 50 0 NA
4 GL-002 Cover glass Experimental 55 1 NA
5 LB-001 LB agar Bacteria 5 0 NA
6 LB-002 LB broth Bacteria 6 1 NA
7 MS-001 MS agar Plant 7 2 NA
8 MS-002 MS broth Plant 7 0 NA
9 MS-003 MS agar(w/ plant hormone) Plant 2 1 NA
10 PI-001 Pasteur pipet Experimental 30 5 10000
11 PI-002 Pipet-AID(10ml) Experimental 45 0 15000
12 PI-003 Pipet-AID(25ml) Experimental 45 5 25000
13 PI-004 Pipet-AID Experimental 2 0 NA
Equipment Glasswear Medium
1 NA NA 70000
2 NA NA 90000
3 NA 20000 NA
4 NA 20000 NA
5 NA NA 55000
6 NA NA 50000
7 NA NA 65000
8 NA NA 60000
9 NA NA 75000
10 NA NA NA
11 NA NA NA
12 NA NA NA
13 100000 NA NAtidyr 라이브러리에 있는 spread를 쓰거나... NA 진짜 보기싫네...
> dcast(df2,Order~Cancel,value.var="Price",sum)
Order 0 1 2 5
1 2 100000 75000 0 0
2 5 55000 0 0 0
3 6 0 50000 0 0
4 7 60000 0 65000 0
5 10 0 0 70000 0
6 20 90000 0 0 0
7 30 0 0 0 10000
8 45 15000 0 0 25000
9 50 20000 0 0 0
10 55 0 20000 0 0dcast를 쓰자. 얘가 대충 피벗테이블 같은 역할이라는 듯... 형식은 dcast(원본 데이터, 행~열에 들어갈 항목, 값으로 들어갈 항목, 적용할 함수).
Summarize
대충 설명만 들어봤을 때는 pandas의 그룹바이가 생각난다.
data <- read.table(header=TRUE, text='
subject sex condition before after change
1 F placebo 10.1 6.9 -3.2
2 F placebo 6.3 4.2 -2.1
3 M aspirin 12.4 6.3 -6.1
4 F placebo 8.1 6.1 -2.0
5 M aspirin 15.2 9.9 -5.3
6 F aspirin 10.9 7.0 -3.9
7 F aspirin 11.6 8.5 -3.1
8 M aspirin 9.5 3.0 -6.5
9 F placebo 11.5 9.0 -2.5
10 M placebo 11.9 11.0 -0.9
11 F aspirin 11.4 8.0 -3.4
12 M aspirin 10.0 4.4 -5.6
13 M aspirin 12.5 5.4 -7.1
14 M placebo 10.6 10.6 0.0
15 M aspirin 9.1 4.3 -4.8
16 F placebo 12.1 10.2 -1.9
17 F placebo 11.0 8.8 -2.2
18 F placebo 11.9 10.2 -1.7
19 M aspirin 9.1 3.6 -5.5
20 M placebo 13.5 12.4 -1.1
21 M aspirin 12.0 7.5 -4.5
22 F placebo 9.1 7.6 -1.5
23 M placebo 9.9 8.0 -1.9
24 F placebo 7.6 5.2 -2.4
25 F placebo 11.8 9.7 -2.1
26 F placebo 11.8 10.7 -1.1
27 F aspirin 10.1 7.9 -2.2
28 M aspirin 11.6 8.3 -3.3
29 F aspirin 11.3 6.8 -4.5
30 F placebo 10.3 8.3 -2.0
')예제 테이블은 이건데... 아마도 임상실험을 상정하고 만든 데이터같다. 임상실험에서는 투약군과 위약군이 나뉜다. 투약군은 약을 투여하는거고 위약군은 가짜 약을 투여하는 군이라고 보면 된다.
비운의 약 TGN1412의 경우 전임상에서는 이상 없었는데 1차 임상시험에서 투약군 6명이 전부 심각한 부작용으로 ICU행이 되어서 개발 중단된 약. (위약군 두 명만 멀쩡했다고...)
> library(plyr)
> cdata=ddply(data,c("sex","condition"),summarise,N=length(change),mean=mean(change),sd=sd(change),se=sd/sqrt(N))
> cdata
sex condition N mean sd se
1 F aspirin 5 -3.420000 0.8642916 0.3865230
2 F placebo 12 -2.058333 0.5247655 0.1514867
3 M aspirin 9 -5.411111 1.1307569 0.3769190
4 M placebo 4 -0.975000 0.7804913 0.3902456
# Run the functions length, mean, and sd on the value of "change" for each group,
# broken down by sex + conditionddply()를 쓰고 싶다면 plyr 라이브러리를... 아직도 안 깔았음???
> cdataNA=ddply(dataNA,c("sex","condition"),summarise,N=sum(!is.na(change)),mean=mean(change,na.rm=TRUE),sd=sd(change,na.rm=TRUE),se=sd/sqrt(N))
> cdataNA
sex condition N mean sd se
1 F aspirin 4 -3.425000 0.9979145 0.4989572
2 F placebo 12 -2.058333 0.5247655 0.1514867
3 M aspirin 7 -5.142857 1.0674848 0.4034713
4 M placebo 3 -1.300000 0.5291503 0.3055050네? 결측값이요? na.rm=TRUE를 주면 된다.
> ddply(dataNA,c("sex","condition"),summarise,N=sum(!is.na(change)),mean=mean(change),sd=sd(change,na.rm=TRUE),se=sd/sqrt(N))
sex condition N mean sd se
1 F aspirin 4 NA 0.9979145 0.4989572
2 F placebo 12 -2.058333 0.5247655 0.1514867
3 M aspirin 7 NA 1.0674848 0.4034713
4 M placebo 3 NA 0.5291503 0.3055050na.rm=TRUE를 안 주면(일단 평균에만 안 줬다) 이렇게 된다. pandas는 skipna가 자동으로 켜져있지만 R은 아님.
> library(doBy)
> cdata=summaryBy(change~sex+condition,data=data,FUN=c(length,mean,sd))
> cdata
sex condition change.length change.mean change.sd
1 F aspirin 5 -3.420000 0.8642916
2 F placebo 12 -2.058333 0.5247655
3 M aspirin 9 -5.411111 1.1307569
4 M placebo 4 -0.975000 0.7804913doBy 라이브러리의 summaryBy()도 쓸 수 있다. 저게 ddply()보다 간결함.
cdataNA <- summaryBy(change ~ sex + condition, data=dataNA,
FUN=c(length2, mean, sd), na.rm=TRUE)결측값도 간결하게 처리해준다.
네? 라이브러리 깔 여건이 안된다고요? 그렇다면 R에 있는 aggregate()를 쓰면 되는데... 쓰다보면 알겠지만 차라리 라이브러리 까는 게 낫다.
> cdata=aggregate(data["subject"],by=data[c("sex","condition")],FUN=length)
> cdata
sex condition subject
1 F aspirin 5
2 M aspirin 9
3 F placebo 12
4 M placebo 4일단 만들고
> names(cdata)[names(cdata)=="subject"]="N"
> cdata
sex condition N
1 F aspirin 5
2 M aspirin 9
3 F placebo 12
4 M placebo 4이름 바꿔주고
> cdata=cdata[order(cdata$sex),]
> cdata
sex condition N
1 F aspirin 5
3 F placebo 12
2 M aspirin 9
4 M placebo 4정렬해주고... 아직 안 끝났다. 이제 평균이랑 표준편차랑 se 구해야된다.
> cdata.means=aggregate(data[c("before","after","change")],by=data[c("sex","condition")],FUN=mean)
> cdata.means
sex condition before after change
1 F aspirin 11.06000 7.640000 -3.420000
2 M aspirin 11.26667 5.855556 -5.411111
3 F placebo 10.13333 8.075000 -2.058333
4 M placebo 11.47500 10.500000 -0.975000> cdata=merge(cdata,cdata.means)
> cdata
sex condition N before after change
1 F aspirin 5 11.06000 7.640000 -3.420000
2 F placebo 12 10.13333 8.075000 -2.058333
3 M aspirin 9 11.26667 5.855556 -5.411111
4 M placebo 4 11.47500 10.500000 -0.975000평균
> cdata.sd=aggregate(data["change"],by=data[c("sex","condition")],FUN=sd)
> cdata.sd
sex condition change
1 F aspirin 0.8642916
2 M aspirin 1.1307569
3 F placebo 0.5247655
4 M placebo 0.7804913> names(cdata.sd)[names(cdata.sd)=="change"] <- "change.sd"
> cdata.sd
sex condition change.sd
1 F aspirin 0.8642916
2 M aspirin 1.1307569
3 F placebo 0.5247655
4 M placebo 0.7804913> cdata=merge(cdata,cdata.sd)
> cdata
sex condition subject before after change change.sd
1 F aspirin 5 11.06000 7.640000 -3.420000 0.8642916
2 F placebo 12 10.13333 8.075000 -2.058333 0.5247655
3 M aspirin 9 11.26667 5.855556 -5.411111 1.1307569
4 M placebo 4 11.47500 10.500000 -0.975000 0.7804913표준편차(Standard deviation 줄이면 SD)
> cdata$change.se <- cdata$change.sd / sqrt(cdata$subject)
> cdata
sex condition subject before after change change.sd change.se
1 F aspirin 5 11.06000 7.640000 -3.420000 0.8642916 0.3865230
2 F placebo 12 10.13333 8.075000 -2.058333 0.5247655 0.1514867
3 M aspirin 9 11.26667 5.855556 -5.411111 1.1307569 0.3769190
4 M placebo 4 11.47500 10.500000 -0.975000 0.7804913 0.3902456se... 저거 참고로 중간에 표 한번 조져서 다시 만들었다...ㅋㅋㅋㅋㅋㅋ
이 노가다를 하느니 그냥 라이브러리를 깔자...
Contingency table
솔직히 이거 뭐하는건지는 나도 모름.
> ctable=table(df)
> ctable
, , Class = Basic
Interested.in
ID Java Python R
ahn_0526 0 0 0
comma_life 1 0 0
hanguk_joa 0 0 0
kimlab0213 0 1 0
kokoatalk 1 0 0
kuda_koma 0 0 0
peponi01 0 0 1
sigma_00 0 0 1
wheresjohn 0 0 0
, , Class = Expert
Interested.in
ID Java Python R
ahn_0526 0 0 0
comma_life 0 0 0
hanguk_joa 0 1 0
kimlab0213 0 0 0
kokoatalk 0 0 0
kuda_koma 0 0 1
peponi01 0 0 0
sigma_00 0 0 0
wheresjohn 0 0 0
, , Class = Medium
Interested.in
ID Java Python R
ahn_0526 0 1 0
comma_life 0 0 0
hanguk_joa 0 0 0
kimlab0213 0 0 0
kokoatalk 0 0 0
kuda_koma 0 0 0
peponi01 0 0 0
sigma_00 0 0 0
wheresjohn 1 0 0일단 불러온 걸로 만들어 본 결과...
> df=read.csv('/home/koreanraichu/example.csv',sep=";")
> df
ID Interested.in Class
1 kimlab0213 Python Basic
2 ahn_0526 Python Medium
3 peponi01 R Basic
4 kuda_koma R Expert
5 comma_life Java Basic
6 wheresjohn Java Medium
7 hanguk_joa Python Expert
8 sigma_00 R Basic
9 kokoatalk Java Basic여기서 Class별로 언어 몇 개 있는지 세 준다. (Basic에서 파이썬 R 자바 몇개 이런 식) 그걸 ID별로 표기하느라 중구난방이 된 것. 이걸 수제작으로 그리면
> df_counts=data.frame(Class=c("Basic","Basic","Basic","Medium","Medium","Medium","Expert","Expert","Expert"),Lan=c("Python","R","Java","Python","R","Java","Python","R","Java"),freq=c(1,2,2,1,0,1,1,1,0))
> df_counts
Class Lan freq
1 Basic Python 1
2 Basic R 2
3 Basic Java 2
4 Medium Python 1
5 Medium R 0
6 Medium Java 1
7 Expert Python 1
8 Expert R 1
9 Expert Java 0중간에 오타나면 멘탈바사삭 확정.
우리의 신입사원 김부추씨는 프로그래밍 언어 강의 플랫폼을 서비스하는 회사에서 근무한다. 회사에서 고객 데이터 집계를 위해 각 클래스별로 얼마나 듣는지 데이터를 달라는데... 저걸 다 세면 되나요? 아니 어느세월에...
> table(df$Class,df$Interested.in)
Java Python R
Basic 2 1 2
Expert 0 1 1
Medium 1 1 0이러면 나오잖음...
인덱스는 난이도(class), 컬럼이 언어이다. 즉 컬럼별로 보자면 자바 셋, 파이썬 셋, R 셋. 난이도별로 보자면 초급 5명, 중급 2명, 쌉고수전문가 2명.
네? 회사에 제출하는건데 저렇게 해도 되냐고요?
> table(df$Class,df$Interested.in,dnn=c("Class","Interested.in"))
Interested.in
Class Java Python R
Basic 2 1 2
Expert 0 1 1
Medium 1 1 0이름을 넣으면 됨.
> table(df[,c("Class","Interested.in")])
Interested.in
Class Java Python R
Basic 2 1 2
Expert 0 1 1
Medium 1 1 0물론 이것도 된다. 저기서 ID를 넣으면 데이터가 매우 괴랄해지므로 좀 묶을 수 있는 걸로 넣어보는 것을 추천한다.
> countdf=as.data.frame(table(df))
> countdf
ID Interested.in Class Freq
1 ahn_0526 Java Basic 0
2 comma_life Java Basic 1
3 hanguk_joa Java Basic 0
4 kimlab0213 Java Basic 0
5 kokoatalk Java Basic 1
6 kuda_koma Java Basic 0
7 peponi01 Java Basic 0
8 sigma_00 Java Basic 0
9 wheresjohn Java Basic 0
10 ahn_0526 Python Basic 0
11 comma_life Python Basic 0
12 hanguk_joa Python Basic 0
13 kimlab0213 Python Basic 1
14 kokoatalk Python Basic 0
15 kuda_koma Python Basic 0
16 peponi01 Python Basic 0
17 sigma_00 Python Basic 0
18 wheresjohn Python Basic 0
19 ahn_0526 R Basic 0
20 comma_life R Basic 0
21 hanguk_joa R Basic 0
22 kimlab0213 R Basic 0
23 kokoatalk R Basic 0
24 kuda_koma R Basic 0
25 peponi01 R Basic 1
26 sigma_00 R Basic 1
27 wheresjohn R Basic 0
28 ahn_0526 Java Expert 0
29 comma_life Java Expert 0
30 hanguk_joa Java Expert 0
31 kimlab0213 Java Expert 0
32 kokoatalk Java Expert 0
33 kuda_koma Java Expert 0
34 peponi01 Java Expert 0
35 sigma_00 Java Expert 0
36 wheresjohn Java Expert 0
37 ahn_0526 Python Expert 0
38 comma_life Python Expert 0
39 hanguk_joa Python Expert 1
40 kimlab0213 Python Expert 0
41 kokoatalk Python Expert 0
42 kuda_koma Python Expert 0
43 peponi01 Python Expert 0
44 sigma_00 Python Expert 0
45 wheresjohn Python Expert 0
46 ahn_0526 R Expert 0
47 comma_life R Expert 0
48 hanguk_joa R Expert 0
49 kimlab0213 R Expert 0
50 kokoatalk R Expert 0
51 kuda_koma R Expert 1
52 peponi01 R Expert 0
53 sigma_00 R Expert 0
54 wheresjohn R Expert 0
55 ahn_0526 Java Medium 0
56 comma_life Java Medium 0
57 hanguk_joa Java Medium 0
58 kimlab0213 Java Medium 0
59 kokoatalk Java Medium 0
60 kuda_koma Java Medium 0
61 peponi01 Java Medium 0
62 sigma_00 Java Medium 0
63 wheresjohn Java Medium 1
64 ahn_0526 Python Medium 1
65 comma_life Python Medium 0
66 hanguk_joa Python Medium 0
67 kimlab0213 Python Medium 0
68 kokoatalk Python Medium 0
69 kuda_koma Python Medium 0
70 peponi01 Python Medium 0
71 sigma_00 Python Medium 0
72 wheresjohn Python Medium 0
73 ahn_0526 R Medium 0
74 comma_life R Medium 0
75 hanguk_joa R Medium 0
76 kimlab0213 R Medium 0
77 kokoatalk R Medium 0
78 kuda_koma R Medium 0
79 peponi01 R Medium 0
80 sigma_00 R Medium 0
81 wheresjohn R Medium 0count로 바꾸는 것도 되긴 되는데... 하지 말자... (마른세수)
> xtabs(Freq~Class+Interested.in,data=countdf)
Interested.in
Class Java Python R
Basic 2 1 2
Expert 0 1 1
Medium 1 1 0뭐야 이것도 됨?
'Coding > R' 카테고리의 다른 글
| R 배워보기-7. Statistical analysis (상) (0) | 2022.08.21 |
|---|---|
| R 배워보기- 6.5. Manipulating data-Sequential data (0) | 2022.08.20 |
| R 배워보기- 6.3. Manipulating data-Data Frames (0) | 2022.08.20 |
| R 배워보기- 6.2. Manipulating data-Factors (0) | 2022.08.20 |
| R 배워보기- 6.1. Manipulating data-General (0) | 2022.08.20 |
들어가기 전에 작은 시범조교를 하나(아니고 넷) 준비했음.. 다운 ㄱㄱ
각 csv파일의 내용물을 R로 불러오면
> df=read.csv('/home/koreanraichu/example.csv',sep=";")
> df
ID Interesred.in Class
1 kimlab0213 Python Basic
2 ahn_0526 Python Medium
3 peponi01 R Basic
4 kuda_koma R Expert
5 comma_life Java Basic
6 wheresjohn Java Medium
7 hanguk_joa Python Expert
8 sigma_00 R Basic
9 kokoatalk Java Basic(example, 구분자 세미콜론)
> df2=read.csv('/home/koreanraichu/example2.csv')
> df2
ID Product.name Category.1 Category.2 Price Order Cancel
1 LB-001 LB agar Medium Bacteria 55000 5 0
2 LB-002 LB broth Medium Bacteria 50000 6 1
3 MS-001 MS agar Medium Plant 65000 7 2
4 MS-002 MS broth Medium Plant 60000 7 0
5 DM-001 DMEM Medium Cell culture 90000 20 0
6 BC-001 BCIG agar Medium Bacteria 70000 10 2
7 MS-003 MS agar(w/ plant hormone) Medium Plant 75000 2 1
8 PI-001 Pasteur pipet Consumable Experimental 10000 30 5
9 PI-002 Pipet-AID(10ml) Consumable Experimental 15000 45 0
10 PI-003 Pipet-AID(25ml) Consumable Experimental 25000 45 5
11 PI-004 Pipet-AID Equipment Experimental 100000 2 0
12 GL-001 Slide glass Glasswear Experimental 20000 50 0
13 GL-002 Cover glass Glasswear Experimental 20000 55 1(example2, 구분자 콤마) 만들어놓고 잊혀진 파일
> df3=read.csv('/home/koreanraichu/example3.csv',sep="\t")
> df3
name Chemical.formula MW.g.mol.1. Density.g.cm.3.
1 Sodium chloride NaCl 58.443 2.17000
2 Acetic acid C2H4O2 60.052 1.04900
3 Glucose C6H12O6 180.156 1.54000
4 Mercury(II) sulfide HgS 232.660 8.10000
5 Toluene C7H8 92.141 0.87000
6 Phenol C6H6O 94.113 1.07000
7 Glycerol C3H8O3 92.094 1.26100
8 PETN C5H8N4O12 316.137 1.77000
9 Ethanol C2H6O 46.069 0.78945
10 SDS C12H25NaSO4 288.372 1.01000
11 Chlorophyll a C55H72MgN4O5 893.509 1.07900
12 Citric acid anhydrous C6H8O7 192.123 1.66500
13 Boron tribromide BBr3 250.520 2.64300(example3, 구분자 탭)
> df4=read.csv('/home/koreanraichu/example4.csv',sep=" ")
> df4
category compound_name chemical_formula Molecular_mass.g.mol.1.
1 Borane Borane BH3 13.830
2 Borane Ammonia_borane BNH6 30.865
3 Borane Tetraborane B4H10 53.320
4 Borane Pentaborane(9) B5H9 63.120
5 Borane Octadecaborane B18H22 216.770
6 Oxide Caesium_monooxide Cs2O 281.810
7 Oxide Actinium_oxide Ac2O3 502.053
8 Oxide Triuranium_oxide U3O8 842.100
9 Oxide Technetium(VII)_oxide Tc2O7 307.810
10 Oxide Thorium_monooxide ThO 248.040
11 Alkane Methane CH4 16.043
12 Alkane Hexane C6H14 86.178
13 Alkane Nonane C9H20 128.259
14 Alkane Tridecane C13H28 184.367
15 Alkane Hentricotane C31H64 436.853
16 Sugar_alcohol Mannotol C6H14O6 182.172
17 Sugar_alcohol Xylitol C5H12O5 152.146
18 Sugar_alcohol Fucitol C6H14O5 166.170
19 Sugar_alcohol Volemitol C7H16O7 212.198
20 Sugar_alcohol Inositol C6H12O6 180.160
21 IARC_group_1 Aflatoxin_B1 C17H12O6 312.277
22 IARC_group_1 Cicloaporin C61H111N11O12 1202.635
23 IARC_group_1 Gallium_arsenide GaAs 144.615
24 IARC_group_1 Melphalan C13H18Cl2N2O2 305.200
25 IARC_group_1 Azothioprine C9H7N7O2S 277.260(example4, 구분자 공백)
받아뒀다가 이거 할 때도 쓰고 판다스 할 때도 쓰세요. 참고로 해당 csv파일은 엑셀로도 열 수는 있지만 일단 만들 때는 지에딧으로 만들었음. (gedit filename.확장자 하면 그걸로 뜹디다)
Python과 R의 데이터프레임
-R은 데이터프레임을 다루기 위해 따로 뭘 깔 필요가 없다. 물론 일부 기능을 위해 plyr 라이브러리를 깔긴 해야 하지만 라이브러리 없이도 할 수는 있다. (python은 일단 판다스부터 깔고 봐야 한다)
-얘도 구분자 지정 안 해주면 default는 ,다. (위에 불러오는 코드를 보면 세미콜론과 탭, 공백은 따로 sep=""옵션을 줬다) 원래 csv의 cs가 comma seperated의 약자.
컬럼명 바꾸기
이번 시범조교는 2번 파일. 해당 파일의 컬럼명은
> names(df2)
[1] "ID" "Product.name" "Category.1" "Category.2" "Price"
[6] "Order" "Cancel"이렇게 되어 있다.
우리의 신입사원 김부추씨는 저 csv파일을 받아서 R로 깔끔하게 정리를 했다. (order는 주문량, cancel은 주문 취소량) 그런데 중간보고를 위해 상급자에게 갔더니 상급자의 한마디!
"부추씨, 이거 정리 되게 깔끔하게 잘 했어. 그런데 저기 ID가 한번에 딱 봐서는 모를 것 같거든... ID만 Product ID로 바꿔서 제출하면 될 것 같아. "
김부추 멘붕왔다. 헐 그럼 csv파일단에서 수정 다시 해야 함? 이거 하느라 개고생했는데...OTL 근데 부추가 누구죠 아 제 텅비드 이름인데요 이제 텅비드를 취업시키는건가 아 걍 해요
> library(plyr)
> rename(df2,c("ID"="Product.ID"))
Product.ID Product.name Category.1 Category.2 Price Order
1 LB-001 LB agar Medium Bacteria 55000 5
2 LB-002 LB broth Medium Bacteria 50000 6
3 MS-001 MS agar Medium Plant 65000 7
4 MS-002 MS broth Medium Plant 60000 7
5 DM-001 DMEM Medium Cell culture 90000 20
6 BC-001 BCIG agar Medium Bacteria 70000 10
7 MS-003 MS agar(w/ plant hormone) Medium Plant 75000 2
8 PI-001 Pasteur pipet Consumable Experimental 10000 30
9 PI-002 Pipet-AID(10ml) Consumable Experimental 15000 45
10 PI-003 Pipet-AID(25ml) Consumable Experimental 25000 45
11 PI-004 Pipet-AID Equipment Experimental 100000 2
12 GL-001 Slide glass Glasswear Experimental 20000 50
13 GL-002 Cover glass Glasswear Experimental 20000 55
Cancel
1 0
2 1
3 2
4 0
5 0
6 2
7 1
8 5
9 0
10 5
11 0
12 0
13 1파일단에서 손댈 것도 없이 그냥 저거 한 방이면 끝난다. (물론 plyr 라이브러리는 깔아두셨죠?) 참고로 얘는 저거 적용하고 names(df2)로 불러보면 ID로 나온다. (df2=코드 하고 해야 하나...)
네? 당장 보고 들어가야 해서 plyr 라이브러리 깔 시간이 없다고요?
> names(df2)[names(df2)=="ID"]="Product.ID"
> df2
Product.ID Product.name Category.1 Category.2 Price Order
1 LB-001 LB agar Medium Bacteria 55000 5
2 LB-002 LB broth Medium Bacteria 50000 6
3 MS-001 MS agar Medium Plant 65000 7
4 MS-002 MS broth Medium Plant 60000 7
5 DM-001 DMEM Medium Cell culture 90000 20
6 BC-001 BCIG agar Medium Bacteria 70000 10
7 MS-003 MS agar(w/ plant hormone) Medium Plant 75000 2
8 PI-001 Pasteur pipet Consumable Experimental 10000 30
9 PI-002 Pipet-AID(10ml) Consumable Experimental 15000 45
10 PI-003 Pipet-AID(25ml) Consumable Experimental 25000 45
11 PI-004 Pipet-AID Equipment Experimental 100000 2
12 GL-001 Slide glass Glasswear Experimental 20000 50
13 GL-002 Cover glass Glasswear Experimental 20000 55
Cancel
1 0
2 1
3 2
4 0
5 0
6 2
7 1
8 5
9 0
10 5
11 0
12 0
13 1ㄱㄱ
> names(df2)=sub("ID","Product.ID",names(df2))
> df2
Product.Product.ID Product.name Category.1 Category.2 Price
1 LB-001 LB agar Medium Bacteria 55000
2 LB-002 LB broth Medium Bacteria 50000
3 MS-001 MS agar Medium Plant 65000
4 MS-002 MS broth Medium Plant 60000
5 DM-001 DMEM Medium Cell culture 90000
6 BC-001 BCIG agar Medium Bacteria 70000
7 MS-003 MS agar(w/ plant hormone) Medium Plant 75000
8 PI-001 Pasteur pipet Consumable Experimental 10000
9 PI-002 Pipet-AID(10ml) Consumable Experimental 15000
10 PI-003 Pipet-AID(25ml) Consumable Experimental 25000
11 PI-004 Pipet-AID Equipment Experimental 100000
12 GL-001 Slide glass Glasswear Experimental 20000
13 GL-002 Cover glass Glasswear Experimental 20000
Order Cancel
1 5 0
2 6 1
3 7 2
4 7 0
5 20 0
6 10 2
7 2 1
8 30 5
9 45 0
10 45 5
11 2 0
12 50 0
13 55 1> names(df2)=gsub("D","d",names(df2))
> df2
Product.Product.Id Product.name Category.1 Category.2 Price
1 LB-001 LB agar Medium Bacteria 55000
2 LB-002 LB broth Medium Bacteria 50000
3 MS-001 MS agar Medium Plant 65000
4 MS-002 MS broth Medium Plant 60000
5 DM-001 DMEM Medium Cell culture 90000
6 BC-001 BCIG agar Medium Bacteria 70000
7 MS-003 MS agar(w/ plant hormone) Medium Plant 75000
8 PI-001 Pasteur pipet Consumable Experimental 10000
9 PI-002 Pipet-AID(10ml) Consumable Experimental 15000
10 PI-003 Pipet-AID(25ml) Consumable Experimental 25000
11 PI-004 Pipet-AID Equipment Experimental 100000
12 GL-001 Slide glass Glasswear Experimental 20000
13 GL-002 Cover glass Glasswear Experimental 20000
Order Cancel
1 5 0
2 6 1
3 7 2
4 7 0
5 20 0
6 10 2
7 2 1
8 30 5
9 45 0
10 45 5
11 2 0
12 50 0
13 55 1sub(), gsub()도 당연히 된다. (라이브러리 없어도 됨)
컬럼 첨삭하기
> df
ID Interesred.in Class
1 kimlab0213 Python Basic
2 ahn_0526 Python Medium
3 peponi01 R Basic
4 kuda_koma R Expert
5 comma_life Java Basic
6 wheresjohn Java Medium
7 hanguk_joa Python Expert
8 sigma_00 R Basic
9 kokoatalk Java Basicexample 1번을 모셔봅시다. 예제 1번은 회원들에게 프로그래밍 언어 강의를 제공하는 웹 플랫폼을 상정하고 만든 것이다. (그래서 파이썬 R 자바가... 씨언어 어디갔어요 씨언어 뭐 씨? 씨샵? 씨쁠쁠?) 그러니까 ID는 회원 아이디, 관심분야는 파이썬, 클래스는 강의의 난이도(초급/중급/전문가).
씁 근데 이렇게만 해서는 이 아이디의 주인이 해당 수업을 다 이수했나 아닌가를 모르겠다. 그래서 P/F 컬럼을 새로 추가하고자 한다.
> df$PF=c("P","P","F","F","P","F","P","F","F")
> df
ID Interesred.in Class PF
1 kimlab0213 Python Basic P
2 ahn_0526 Python Medium P
3 peponi01 R Basic F
4 kuda_koma R Expert F
5 comma_life Java Basic P
6 wheresjohn Java Medium F
7 hanguk_joa Python Expert P
8 sigma_00 R Basic F
9 kokoatalk Java Basic F제목이요? 저거는 따옴표로 감싸지 않는 이상 공백 있으면 에러뜸미다... (언더바 ㄱㄱ)
> df[,"Pass or Fail"]=c("P","P","F","F","P","F","P","F","F")
> df
ID Interesred.in Class Pass or Fail
1 kimlab0213 Python Basic P
2 ahn_0526 Python Medium P
3 peponi01 R Basic F
4 kuda_koma R Expert F
5 comma_life Java Basic P
6 wheresjohn Java Medium F
7 hanguk_joa Python Expert P
8 sigma_00 R Basic F
9 kokoatalk Java Basic F아니면 이건 어떠심?
그렇게 아이디어를 제안한 김상추씨. 하지만 선배 김양상추씨(둘 다 본인 포켓몬 이름임... 릴리요였나...)는 그 제안을 듣고 이렇게 말했다.
"좋은 아이디어긴 한데... P/F로 매기게 되면 강의 이수여부는 알 수 있지만, 출결 관리까지 알기는 좀 힘들 것 같아. 차라리 P/F를 빼고 출석율을 넣거나, 출석율이랑 병기하는 건 어때? "
> df$PF=NULL
> df
ID Interesred.in Class
1 kimlab0213 Python Basic
2 ahn_0526 Python Medium
3 peponi01 R Basic
4 kuda_koma R Expert
5 comma_life Java Basic
6 wheresjohn Java Medium
7 hanguk_joa Python Expert
8 sigma_00 R Basic
9 kokoatalk Java Basic그래서 김상추씨는 PF 컬럼을 빼버렸다.
참고로
# Ways to add a column
data$size <- c("small", "large", "medium")
data[["size"]] <- c("small", "large", "medium")
data[,"size"] <- c("small", "large", "medium")
data$size <- 0 # Use the same value (0) for all rows# Ways to remove the column
data$size <- NULL
data[["size"]] <- NULL
data[,"size"] <- NULL
data[[3]] <- NULL
data[,3] <- NULL
data <- subset(data, select=-size)이건 뭐 니들이 뭘 좋아할 지 몰라서 다 넣었어도 아니고 첨삭 방법이 되게 많다.
컬럼 오더 바꾸기
이번 시범조교...
> df3
name Chemical.formula MW.g.mol.1. Density.g.cm.3.
1 Sodium chloride NaCl 58.443 2.17000
2 Acetic acid C2H4O2 60.052 1.04900
3 Glucose C6H12O6 180.156 1.54000
4 Mercury(II) sulfide HgS 232.660 8.10000
5 Toluene C7H8 92.141 0.87000
6 Phenol C6H6O 94.113 1.07000
7 Glycerol C3H8O3 92.094 1.26100
8 PETN C5H8N4O12 316.137 1.77000
9 Ethanol C2H6O 46.069 0.78945
10 SDS C12H25NaSO4 288.372 1.01000
11 Chlorophyll a C55H72MgN4O5 893.509 1.07900
12 Citric acid anhydrous C6H8O7 192.123 1.66500
13 Boron tribromide BBr3 250.520 2.64300이분임...
보기만 해도 으아아가 절로 나온다면 정상입니다. 난 아니지만. 이사람 집에 주기율표 있다 끝말잇기 잘하시겠네요 아뇨 그건 아닙니다 원래 주기율표 다 꿰고 있으면 완급조절도 가능하다 코페르니슘! 슘페터! 터븀!
> df3[c(4,3,2,1)]
Density.g.cm.3. MW.g.mol.1. Chemical.formula name
1 2.17000 58.443 NaCl Sodium chloride
2 1.04900 60.052 C2H4O2 Acetic acid
3 1.54000 180.156 C6H12O6 Glucose
4 8.10000 232.660 HgS Mercury(II) sulfide
5 0.87000 92.141 C7H8 Toluene
6 1.07000 94.113 C6H6O Phenol
7 1.26100 92.094 C3H8O3 Glycerol
8 1.77000 316.137 C5H8N4O12 PETN
9 0.78945 46.069 C2H6O Ethanol
10 1.01000 288.372 C12H25NaSO4 SDS
11 1.07900 893.509 C55H72MgN4O5 Chlorophyll a
12 1.66500 192.123 C6H8O7 Citric acid anhydrous
13 2.64300 250.520 BBr3 Boron tribromide오더를 이렇게 컬럼명으로 바꾸거나(...)
> df3[c("Chemical.formula","name","MW.g.mol.1.","Density.g.cm.3.")]
Chemical.formula name MW.g.mol.1. Density.g.cm.3.
1 NaCl Sodium chloride 58.443 2.17000
2 C2H4O2 Acetic acid 60.052 1.04900
3 C6H12O6 Glucose 180.156 1.54000
4 HgS Mercury(II) sulfide 232.660 8.10000
5 C7H8 Toluene 92.141 0.87000
6 C6H6O Phenol 94.113 1.07000
7 C3H8O3 Glycerol 92.094 1.26100
8 C5H8N4O12 PETN 316.137 1.77000
9 C2H6O Ethanol 46.069 0.78945
10 C12H25NaSO4 SDS 288.372 1.01000
11 C55H72MgN4O5 Chlorophyll a 893.509 1.07900
12 C6H8O7 Citric acid anhydrous 192.123 1.66500
13 BBr3 Boron tribromide 250.520 2.64300...일단 저건 숫자가 더 효율적임.
> df3[,c(1,2,4,3)]
name Chemical.formula Density.g.cm.3. MW.g.mol.1.
1 Sodium chloride NaCl 2.17000 58.443
2 Acetic acid C2H4O2 1.04900 60.052
3 Glucose C6H12O6 1.54000 180.156
4 Mercury(II) sulfide HgS 8.10000 232.660
5 Toluene C7H8 0.87000 92.141
6 Phenol C6H6O 1.07000 94.113
7 Glycerol C3H8O3 1.26100 92.094
8 PETN C5H8N4O12 1.77000 316.137
9 Ethanol C2H6O 0.78945 46.069
10 SDS C12H25NaSO4 1.01000 288.372
11 Chlorophyll a C55H72MgN4O5 1.07900 893.509
12 Citric acid anhydrous C6H8O7 1.66500 192.123
13 Boron tribromide BBr3 2.64300 250.520뭔 매트릭스 매기듯이 이렇게도 한다.
> df3[2]
Chemical.formula
1 NaCl
2 C2H4O2
3 C6H12O6
4 HgS
5 C7H8
6 C6H6O
7 C3H8O3
8 C5H8N4O12
9 C2H6O
10 C12H25NaSO4
11 C55H72MgN4O5
12 C6H8O7
13 BBr32열을 뽑아봤습니다.
> df3[,2]
[1] NaCl C2H4O2 C6H12O6 HgS C7H8
[6] C6H6O C3H8O3 C5H8N4O12 C2H6O C12H25NaSO4
[11] C55H72MgN4O5 C6H8O7 BBr3
13 Levels: BBr3 C12H25NaSO4 C2H4O2 C2H6O C3H8O3 C55H72MgN4O5 ... NaCl이렇게 하면 벡터처럼 뽑힌다. (레벨이 있는 걸 보면 아시겠지만 팩터임)
> df3[,2,drop=FALSE]
Chemical.formula
1 NaCl
2 C2H4O2
3 C6H12O6
4 HgS
5 C7H8
6 C6H6O
7 C3H8O3
8 C5H8N4O12
9 C2H6O
10 C12H25NaSO4
11 C55H72MgN4O5
12 C6H8O7
13 BBr3drop=FALSE를 주면 표 형태로 뽑힌다. 저게 그 시리즌가 그거냐 그건 판다스고
파이널 퓨전
> df
ID Interesred.in Class Pass or Fail
1 kimlab0213 Python Basic P
2 ahn_0526 Python Medium P
3 peponi01 R Basic F
4 kuda_koma R Expert F
5 comma_life Java Basic P
6 wheresjohn Java Medium F
7 hanguk_joa Python Expert P
8 sigma_00 R Basic F
9 kokoatalk Java Basic F아까 그거 맞다. 여기에다가 클래스별 가격 정보를 추가할건데... 그럼 가격표가 어디 있느냐고?
> df5=read.table(header=TRUE, text='
+ Class;Price
+ Basic;Free
+ Medium;1000
+ Expert;2000
+ ',sep=";")> df5
Class Price
1 Basic Free
2 Medium 1000
3 Expert 2000여기요.
이 표를 파이널(별)퓨전해달라는 업무를 받은 김상추씨(아니 진짜 내 포켓몬 이름이여 이름이 왜그래요 부추때문에 추로 끝나는 야채는 다 넣었음 다행히도 고추는 없습니다 그건 어감이 거시기해서)... 아니 그럼 새로 컬럼 만들고 저걸 일일이 다 써요? ㄴㄴ
> merge(df,df5,"Class")
Class ID Interesred.in Pass or Fail Price
1 Basic kimlab0213 Python P Free
2 Basic peponi01 R F Free
3 Basic sigma_00 R F Free
4 Basic comma_life Java P Free
5 Basic kokoatalk Java F Free
6 Expert hanguk_joa Python P 2000
7 Expert kuda_koma R F 2000
8 Medium ahn_0526 Python P 1000
9 Medium wheresjohn Java F 1000공통된 컬럼인 Class를 기반으로 파이널퓨전 하면 된다.
> df6[c(3,4,5,1,2)]
ID Interesred.in Pass or Fail Class Price
1 kimlab0213 Python P Basic Free
2 peponi01 R F Basic Free
3 sigma_00 R F Basic Free
4 comma_life Java P Basic Free
5 kokoatalk Java F Basic Free
6 hanguk_joa Python P Expert 2000
7 kuda_koma R F Expert 2000
8 ahn_0526 Python P Medium 1000
9 wheresjohn Java F Medium 1000새 데이터프레임에 할당하고 순서 바꿀 수 있다.
> stories2 <- read.table(header=TRUE, text='
+ id title
+ 1 lions
+ 2 tigers
+ 3 bears
+ ')
subject storyid rating
1 1 1 6.7
2 1 2 4.5
3 1 3 3.7
4 2 2 3.3
5 2 3 4.1
6 2 1 5.2컬럼명이 다른데 되나요?
> merge(x=stories2,y=data,by.x="id",by.y="storyid")
id title subject rating
1 1 lions 1 6.7
2 1 lions 2 5.2
3 2 tigers 1 4.5
4 2 tigers 2 3.3
5 3 bears 1 3.7
6 3 bears 2 4.1네. 근데 내가 갖고 있는거에 만들어서 할랬더니 에러뜸...
Error in fix.by(by.x, x) : 'by' must specify a uniquely valid column혹시 이 에러 해결법 아시는 분 제보좀 부탁드립니다. (합치려는 데이터프레임 영역 팩터로 같은 거 확인함)
> animals <- read.table(header=T, text='
+ size type name
+ small cat lynx
+ big cat tiger
+ small dog chihuahua
+ big dog "great dane"
+ ')
> observations <- read.table(header=T, text='
+ number size type
+ 1 big cat
+ 2 small dog
+ 3 small dog
+ 4 big dog
+ ')이걸 파이널퓨전하는 것도 된다.
> merge(observations,animals,c("size","type"))
size type number name
1 big cat 1 tiger
2 big dog 4 great dane
3 small dog 2 chihuahua
4 small dog 3 chihuahua어째서인지는 모르겠으나 됨.
데이터프레임 비교하기
> dfA <- data.frame(Subject=c(1,1,2,2), Response=c("X","X","X","X"))
> dfB <- data.frame(Subject=c(1,2,3), Response=c("X","Y","X"))
> dfC <- data.frame(Subject=c(1,2,3), Response=c("Z","Y","Z"))
> dfA
Subject Response
1 1 X
2 1 X
3 2 X
4 2 X
> dfB
Subject Response
1 1 X
2 2 Y
3 3 X
> dfC
Subject Response
1 1 Z
2 2 Y
3 3 Z쿡북에 있는 시범조교를 모셔봤습니다.
> dfA$Coder="A"
> dfB$Coder="B"
> dfC$Coder="C"각 데이터프레임에 Coder라는 컬럼을 추가한다.
> df7=rbind(dfA,dfB,dfC)
> df7
Subject Response Coder
1 1 X A
2 1 X A
3 2 X A
4 2 X A
5 1 X B
6 2 Y B
7 3 X B
8 1 Z C
9 2 Y C
10 3 Z C그리고 행단위로 파이널퓨전!
> df7=df7[,c("Coder","Subject","Response")]
> df7
Coder Subject Response
1 A 1 X
2 A 1 X
3 A 2 X
4 A 2 X
5 B 1 X
6 B 2 Y
7 B 3 X
8 C 1 Z
9 C 2 Y
10 C 3 Z하고 정렬까지 해야 시범조교 끝...
참고로 이걸 진행하려면 함수 하나를 정의하고 가야 하는데
dupsBetweenGroups <- function (df, idcol) {
# df: the data frame
# idcol: the column which identifies the group each row belongs to
# Get the data columns to use for finding matches
datacols <- setdiff(names(df), idcol)
# Sort by idcol, then datacols. Save order so we can undo the sorting later.
sortorder <- do.call(order, df)
df <- df[sortorder,]
# Find duplicates within each id group (first copy not marked)
dupWithin <- duplicated(df)
# With duplicates within each group filtered out, find duplicates between groups.
# Need to scan up and down with duplicated() because first copy is not marked.
dupBetween = rep(NA, nrow(df))
dupBetween[!dupWithin] <- duplicated(df[!dupWithin,datacols])
dupBetween[!dupWithin] <- duplicated(df[!dupWithin,datacols], fromLast=TRUE) | dupBetween[!dupWithin]
# ============= Replace NA's with previous non-NA value ==============
# This is why we sorted earlier - it was necessary to do this part efficiently
# Get indexes of non-NA's
goodIdx <- !is.na(dupBetween)
# These are the non-NA values from x only
# Add a leading NA for later use when we index into this vector
goodVals <- c(NA, dupBetween[goodIdx])
# Fill the indices of the output vector with the indices pulled from
# these offsets of goodVals. Add 1 to avoid indexing to zero.
fillIdx <- cumsum(goodIdx)+1
# The original vector, now with gaps filled
dupBetween <- goodVals[fillIdx]
# Undo the original sort
dupBetween[sortorder] <- dupBetween
# Return the vector of which entries are duplicated across groups
return(dupBetween)
}이놈이다. 정의하고 가자.
> dupRows=dupsBetweenGroups(df7,"Coder")
> dupRows
[1] TRUE TRUE FALSE FALSE TRUE TRUE FALSE FALSE TRUE FALSE근데 이렇게만 해 두면 뭔지 모르것다...
> cbind(df7,dup=dupRows)
Coder Subject Response dup
1 A 1 X TRUE
2 A 1 X TRUE
3 A 2 X FALSE
4 A 2 X FALSE
5 B 1 X TRUE
6 B 2 Y TRUE
7 B 3 X FALSE
8 C 1 Z FALSE
9 C 2 Y TRUE
10 C 3 Z FALSE열단위로 결합해도 뭔지 모르겠다...
사실 저건 각 테이블에서 중복되는 값을 찾아주는 함수이다. 코드가 달라도 중복되는 값이 있으면 TRUE, 아예 없으면 FALSE.
> cbind(df7,unique=!dupRows)
Coder Subject Response unique
1 A 1 X FALSE
2 A 1 X FALSE
3 A 2 X TRUE
4 A 2 X TRUE
5 B 1 X FALSE
6 B 2 Y FALSE
7 B 3 X TRUE
8 C 1 Z TRUE
9 C 2 Y FALSE
10 C 3 Z TRUE이건 반대로 중복이 없으면 TRUE, 중복값이 있으면 FALSE다.
서브셋 나누기
> dfA=subset(dfDup,Coder=="A",select=-Coder)
> dfB=subset(dfDup,Coder=="B",select=-Coder)
> dfC=subset(dfDup,Coder=="C",select=-Coder)
> dfA
Subject Response dup
1 1 X TRUE
2 1 X TRUE
3 2 X FALSE
4 2 X FALSE
> dfB
Subject Response dup
5 1 X TRUE
6 2 Y TRUE
7 3 X FALSE
> dfC
Subject Response dup
8 1 Z FALSE
9 2 Y TRUE
10 3 Z FALSE
# 아 저게 Coder 빼고 셀렉션해달라는 얘기였냐고나누면 됩니다. 근데 저 셀렉트는 뭐냐...
> df4
category compound_name chemical_formula Molecular_mass.g.mol.1.
1 Borane Borane BH3 13.830
2 Borane Ammonia_borane BNH6 30.865
3 Borane Tetraborane B4H10 53.320
4 Borane Pentaborane(9) B5H9 63.120
5 Borane Octadecaborane B18H22 216.770
6 Oxide Caesium_monooxide Cs2O 281.810
7 Oxide Actinium_oxide Ac2O3 502.053
8 Oxide Triuranium_oxide U3O8 842.100
9 Oxide Technetium(VII)_oxide Tc2O7 307.810
10 Oxide Thorium_monooxide ThO 248.040
11 Alkane Methane CH4 16.043
12 Alkane Hexane C6H14 86.178
13 Alkane Nonane C9H20 128.259
14 Alkane Tridecane C13H28 184.367
15 Alkane Hentricotane C31H64 436.853
16 Sugar_alcohol Mannotol C6H14O6 182.172
17 Sugar_alcohol Xylitol C5H12O5 152.146
18 Sugar_alcohol Fucitol C6H14O5 166.170
19 Sugar_alcohol Volemitol C7H16O7 212.198
20 Sugar_alcohol Inositol C6H12O6 180.160
21 IARC_group_1 Aflatoxin_B1 C17H12O6 312.277
22 IARC_group_1 Cicloaporin C61H111N11O12 1202.635
23 IARC_group_1 Gallium_arsenide GaAs 144.615
24 IARC_group_1 Melphalan C13H18Cl2N2O2 305.200
25 IARC_group_1 Azothioprine C9H7N7O2S 277.260시범조교 4번을 불러보자. 으아아 저게 뭐야 참고로 IARC group 1은 이놈은 Carcinogen(발암물질)이 확실하니까 먹지도 마시지도 말고 애비 지지해야되는 것들.
> df4A=subset(df4,category=="Borane")
> df4A
category compound_name chemical_formula Molecular_mass.g.mol.1.
1 Borane Borane BH3 13.830
2 Borane Ammonia_borane BNH6 30.865
3 Borane Tetraborane B4H10 53.320
4 Borane Pentaborane(9) B5H9 63.120
5 Borane Octadecaborane B18H22 216.770Borane(붕소 화합물임... 자세한건 모름)으로 서브셋을 만들어보면 이렇게 나온다. 근데 생각해보니 Borane으로 묶었는데 저기서 카테고리를 보여 줄 필요가 없거든.
> df4B=subset(df4,category=="IARC_group_1",select=-category)
> df4B
compound_name chemical_formula Molecular_mass.g.mol.1.
21 Aflatoxin_B1 C17H12O6 312.277
22 Cicloaporin C61H111N11O12 1202.635
23 Gallium_arsenide GaAs 144.615
24 Melphalan C13H18Cl2N2O2 305.200
25 Azothioprine C9H7N7O2S 277.260그래서 select에 -를 줘서 뺀 것이다. (R에서 -붙으면 그거 빼고 달라는 얘기)
> df4C=subset(df4,Molecular_mass.g.mol.1.>=100)
> df4C
category compound_name chemical_formula Molecular_mass.g.mol.1.
5 Borane Octadecaborane B18H22 216.770
6 Oxide Caesium_monooxide Cs2O 281.810
7 Oxide Actinium_oxide Ac2O3 502.053
8 Oxide Triuranium_oxide U3O8 842.100
9 Oxide Technetium(VII)_oxide Tc2O7 307.810
10 Oxide Thorium_monooxide ThO 248.040
13 Alkane Nonane C9H20 128.259
14 Alkane Tridecane C13H28 184.367
15 Alkane Hentricotane C31H64 436.853
16 Sugar_alcohol Mannotol C6H14O6 182.172
17 Sugar_alcohol Xylitol C5H12O5 152.146
18 Sugar_alcohol Fucitol C6H14O5 166.170
19 Sugar_alcohol Volemitol C7H16O7 212.198
20 Sugar_alcohol Inositol C6H12O6 180.160
21 IARC_group_1 Aflatoxin_B1 C17H12O6 312.277
22 IARC_group_1 Cicloaporin C61H111N11O12 1202.635
23 IARC_group_1 Gallium_arsenide GaAs 144.615
24 IARC_group_1 Melphalan C13H18Cl2N2O2 305.200
25 IARC_group_1 Azothioprine C9H7N7O2S 277.260참고로 조건부로 서브셋 만드는 것도 된다. 뭐 여기서는 100으로 잡았지만 ChEMBL에서 불러온 다음 RO5 조건 만족하는 것들로 만들거나 BBB 통과 조건으로 만들거나 할 수도 있다. (아니면 SMILES에 @@ 붙은거?)
팩터 레벨 재조정하기
그 뮤츠씨가 좋아하는? 아니 그건 팩트라니까 얘는 팩터잖아
> str(df3)
'data.frame': 13 obs. of 4 variables:
$ name : Factor w/ 13 levels "Acetic acid",..: 12 1 6 8 13 10 7 9 5 11 ...
$ Chemical.formula: Factor w/ 13 levels "BBr3","C12H25NaSO4",..: 13 3 8 12 11 9 5 7 4 2 ...
$ MW.g.mol.1. : num 58.4 60.1 180.2 232.7 92.1 ...
$ Density.g.cm.3. : num 2.17 1.05 1.54 8.1 0.87 ...3번 시범조교에도 팩터가 두 개 있다. (이름, 분자식) ㅇㅋ? ㄱㄱ
> df3_1=droplevels(df3)
> df3_1
name Chemical.formula MW.g.mol.1. Density.g.cm.3.
1 Sodium chloride NaCl 58.443 2.17000
2 Acetic acid C2H4O2 60.052 1.04900
3 Glucose C6H12O6 180.156 1.54000
4 Mercury(II) sulfide HgS 232.660 8.10000
5 Toluene C7H8 92.141 0.87000
6 Phenol C6H6O 94.113 1.07000
7 Glycerol C3H8O3 92.094 1.26100
8 PETN C5H8N4O12 316.137 1.77000
9 Ethanol C2H6O 46.069 0.78945
10 SDS C12H25NaSO4 288.372 1.01000
11 Chlorophyll a C55H72MgN4O5 893.509 1.07900
13 Boron tribromide BBr3 250.520 2.6430012행을 날리고 droplevel()을 적용하면
> str(df3_1)
'data.frame': 12 obs. of 4 variables:
$ name : Factor w/ 12 levels "Acetic acid",..: 11 1 5 7 12 9 6 8 4 10 ...
$ Chemical.formula: Factor w/ 12 levels "BBr3","C12H25NaSO4",..: 12 3 8 11 10 9 5 7 4 2 ...
$ MW.g.mol.1. : num 58.4 60.1 180.2 232.7 92.1 ...
$ Density.g.cm.3. : num 2.17 1.05 1.54 8.1 0.87 ...팩터 레벨이 바뀌었다. (13->12)
이걸 vapply()와 lapply()를 적용해서 할 수도 있는데
> factor_cols=vapply(df3,is.factor,logical(1))
> factor_cols
name Chemical.formula MW.g.mol.1. Density.g.cm.3.
TRUE TRUE FALSE FALSEvapply()를 통해 이놈이 팩터인가를 볼 수 있고
> df3[factor_cols]=lapply(df3[factor_cols],factor)
# Apply the factor() function to those columns, and assign then back into d팩터 함수를 적용시키면
> str(df3)
'data.frame': 12 obs. of 4 variables:
$ name : Factor w/ 12 levels "Acetic acid",..: 11 1 5 7 12 9 6 8 4 10 ...
$ Chemical.formula: Factor w/ 12 levels "BBr3","C12H25NaSO4",..: 12 3 8 11 10 9 5 7 4 2 ...
$ MW.g.mol.1. : num 58.4 60.1 180.2 232.7 92.1 ...
$ Density.g.cm.3. : num 2.17 1.05 1.54 8.1 0.87 ...(대충 droplevel()이랑 같은거라는 얘기)
'Coding > R' 카테고리의 다른 글
| R 배워보기- 6.5. Manipulating data-Sequential data (0) | 2022.08.20 |
|---|---|
| R 배워보기- 6.4. Manipulating data-Restructing data (0) | 2022.08.20 |
| R 배워보기- 6.2. Manipulating data-Factors (0) | 2022.08.20 |
| R 배워보기- 6.1. Manipulating data-General (0) | 2022.08.20 |
| R 배워보기-5. 데이터 불러오고 쓰기 (0) | 2022.08.20 |
R의 데이터에는 벡터와 팩터가 있다. 그리고 숫자벡터-문자벡터-팩터간에 변환이 가능하다. 어쨌든 가능함.
팩터란 무엇인가
뮤츠씨가 좋아하는거 그건 팩트고 아무튼 벡터와 달리 팩터를 단식으로 뽑게 되면 한 가지 요소가 더 나오게 된다. 그것이 바로 '레벨'이다.
> v=factor(c("A","B","C","D","E","F"))
> v
[1] A B C D E F
Levels: A B C D E F> w=factor(c("35S Promoter","pHellsgate","pStargate","pWatergate","pHellsgate"))
> w
[1] 35S Promoter pHellsgate pStargate pWatergate pHellsgate
Levels: 35S Promoter pHellsgate pStargate pWatergate팩터는 안에 들어있는 요소들로 레벨을 결정한다. 이 때 중복되는 원소는 레벨링에서 빠진다. 선생님 질문있는데 헬게이트 앞에 왜 p가 있나요 그거 벡터임 예? 벡터라고 왜죠 그건 모르는디?
참고: https://www.addgene.org/vector-database/5976/
Addgene: Vector Database - pHELLSGATE 12
This vector is NOT available from Addgene.
www.addgene.org
진짜로 있는 벡터임
https://www.csiro.au/en/work-with-us/services/sample-procurement/rnai-material-transfer-agreement
Hairpin RNAi vectors for plants - Material Transfer Agreement - CSIRO
CSIRO acknowledges the Traditional Owners of the land, sea and waters, of the area that we live and work on across Australia. We acknowledge their continuing connection to their culture and pay our respects to their Elders past and present. View our vision
www.csiro.au
스타게이트랑 워터게이트는 벡터 DB에는 없고 관련된 논문(이거 썼다 이런 논문)이 나오는데 셋 다 RNAi 관련된 transformation 벡터임다.
그니까 내 포켓몬 파티가 난천을 다 깨고 나니 레벨이 60, 61, 59, 60, 62, 60이면 이걸 팩터로 만들었을 때 레벨은 59, 60, 61, 62가 된다. 저기 62짜리는 뭐죠 한카리아스 잡았나요 킹갓키스가 원탑플레이 했나보지
레벨 바꾸기
팩터 레벨이 파이썬 튜플마냥 불변이 아니다. 그래서 바꿀 수 있는데... 여기서도 plyr 라이브러리가 쓰인다.
> library(plyr)
> revalue(v,c("A"="A+"))
[1] A+ B C D E F
Levels: A+ B C D E F> mapvalues(w,from=c("35S Promoter"),to=c("pH2GW7"))
[1] pH2GW7 pHellsgate pStargate pWatergate pHellsgate
Levels: pH2GW7 pHellsgate pStargate pWatergateplyr 라이브러리를 쓰게 되면 revalue()와 mapvalues()를 쓰면 된다.
> levels(v)[levels(v)=="B"]="B+"
> v
[1] A B+ C D E F
Levels: A B+ C D E F> levels(v)[1]="A+"
> v
[1] A+ B+ C D E F
Levels: A+ B+ C D E F> levels(v)=c("A+","B+","C+","D+","E","F")
> v
[1] A+ B+ C+ D+ E F
Levels: A+ B+ C+ D+ E F라이브러리 없이도 이런 방식으로 바꿀 수 있다. 팩터 레벨은 벡터를 써서 뭉텅이로 바꾸는 게 된다.
sub()과 gsub()
사실 둘이 뭔 차인지 나도 모름... 그래서 해봤음.
> v=factor(c("alpha","beta","gamma","alpha","beta"))
> levels(v)=sub("^alpha$","psi",levels(v))
> v
[1] psi beta gamma psi beta
Levels: psi beta gamma> levels(v)=sub("m","M",levels(v))
> v
[1] pI beta gaMma pI beta
Levels: pI beta gaMmasub()은 전체 원소를 치환해주는 건 같지만, 특정 원소의 특정 글자를 치환할 때 첫 글자만 바꿔준다.
> levels(v)=gsub("psi","pi",levels(v))
> v
[1] pi beta gamma pi beta
Levels: pi beta gamma> levels(v)=gsub("m","M",levels(v))
> v
[1] alpha beta gaMMa alpha beta
Levels: alpha beta gaMMagsub()은 sub()과 달리 특정 원소의 모든 특정 글자를 바꿔준다.
레벨 조정-없는 레벨 지우기
> v=factor(c("grass","water","grass"),levels=c("grass","water","fire"))
> v
[1] grass water grass
Levels: grass water fire여기 팩터가 있다. 근데 요소들을 보다 보니... 레벨은 있는데 요소가 없는 게 있네?
> v=factor(v)
> v
[1] grass water grass
Levels: grass water그래서 지워드렸습니다.
> x=factor(c("A","B","A"),levels=c("A","B","C"))
> y=c(1,2,3)
> z=factor(c("R","G","G"),levels=c("R","G","B"))
> df=data.frame(x,y,z)
> df
x y z
1 A 1 R
2 B 2 G
3 A 3 G팩터도 당연히 데이터프레임이 된다. 데이터프레임으로 만들 경우, 데이터프레임으로 출력할 때는 그냥 표로 나오게 되지만
> df$x
[1] A B A
Levels: A B C여기서 팩터 단식으로 불러내면 이렇게 레벨이 나온다.
그런데 여기서도 요소가 없는 레벨이 있다?
> df=droplevels(df)
> df
x y z
1 A 1 R
2 B 2 G
3 A 3 G> df$x
[1] A B A
Levels: A Bdropevels()를 쓰면 지워진다.
레벨 조정-순서 조정
> v=factor(c("S","M","L","XL","M","L"))
> v
[1] S M L XL M L
Levels: L M S XL퍼 펌킨인쨔응 하악하악 뭐여 아무튼... 호바귀와 펌킨인쨔응은 사이즈라는 개념이 있다. 근데 레벨을 보면... 저 순서가 아니다. 알파벳순 아니라고...OTL
> v=factor(v,levels=c("S","M","L","XL"))
> v
[1] S M L XL M L
Levels: S M L XL그래서 바꿔드렸습니다^^
이거 말고도 방법은 많다.
> w=factor(c("pokeball","superball","ultraball","masterball","pokeball"))
> w
[1] pokeball superball ultraball masterball pokeball
Levels: masterball pokeball superball ultraball> w=ordered(c("pokeball","superball","ultraball","masterball"))
> w
[1] pokeball superball ultraball masterball
Levels: masterball < pokeball < superball < ultraballordered()를 써서 정렬하던가... 근데 이걸 이렇게 정렬하면 안되는데...?
> w=factor(c("pokeball","superball","ultraball","pokeball","masterball"),levels=c("pokeball","superball","ultraball","masterball"))
> w
[1] pokeball superball ultraball pokeball masterball
Levels: pokeball superball ultraball masterball만들 때 순서를 아예 정하던가...
> w=factor(c("pokeball","superball","ultraball","masterball","pokeball"))
> w=relevel(w,"masterball")
> w
[1] pokeball superball ultraball masterball pokeball
Levels: masterball pokeball superball ultraball> w=relevel(w,"pokeball")
> w
[1] pokeball superball ultraball masterball pokeball
Levels: pokeball masterball superball ultraball순서가 다 좋은데 앞에 딱 하나만 걸려 그러면 relevel()로 그 걸리는 걸 앞으로 빼버리면 된다.
> x=factor(w,levels=rev(levels(w)))
> x
[1] pokeball superball ultraball pokeball masterball
Levels: masterball ultraball superball pokeball아예 거꾸로 하고 싶을 때는 rev()를 쓰면 된다.
'Coding > R' 카테고리의 다른 글
| R 배워보기- 6.4. Manipulating data-Restructing data (0) | 2022.08.20 |
|---|---|
| R 배워보기- 6.3. Manipulating data-Data Frames (0) | 2022.08.20 |
| R 배워보기- 6.1. Manipulating data-General (0) | 2022.08.20 |
| R 배워보기-5. 데이터 불러오고 쓰기 (0) | 2022.08.20 |
| 번외편-R로 미적분 하기 (0) | 2022.08.20 |
이거 쿡복 보니까 시리즈가 개 많고... 분량이 그냥 종류별로 있습니다... 농담같지만 실화임.
그래서 세부적으로 나갈거예요... 근데 데이터프레임에 csv 불러오는 건 생각 좀 해봐야겠음. 분량이 정말 살인적입니다. 농담 아님.
아 참고로 데이터프레임 정리하기에 라이브러리가 하나 필요합니다. plyr이라고...
그거 없이 하는 방법도 있긴 한데 나중에 뭉텅이로 처리하려면 plyr 필요해요.
install.packages("plyr")설치 ㄱㄱ.
sort()
벡터는 sort()로 정렬한다. 그죠 여기 벡터가 나온다는 건 데이터프레임도 있다 이겁니다. (스포일러)
> v=sample(1:25)
> v
[1] 11 2 12 18 23 21 22 14 3 19 13 9 1 16 5 20 6 10 25 8 4 15 24 7 17지쟈쓰 붓다 알라신을 부르게 만드는 이 데이터를 깔끔하게 정리했으면 좋겠다 그죠?
> sort(v)
[1] 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
> sort(v,decreasing=TRUE)
[1] 25 24 23 22 21 20 19 18 17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1해(별)결. decreasing=TRUE는 파이썬 판다스에서 ascending=T, F 주는것과 비슷하다. 저기에 트루가 들어가면 내림차순. (ascending은... 뭐더라...)
plyr library 소환해서 정리하기
> library(plyr)
> arrange(data,Molecular.Weight)분자량 순으로 정렬한 것. 참고로 터미널에서 부르다보니 분량이 장난없어서 캡처고 뭐고 복사부터 안된다.
> arrange(data,Molecular.Weight,HBA)이건 분자량과 수소결합 받개로 정렬한다는 얘기.
> arrange(data,-Type)> arrange(data,Smiles,-HBA)역방향 정렬은 이런 식으로 -를 붙여서 하면 된다.
상여자는 라이브러리따원 깔지 않는다네
> data[order(data$Name),]단식(이름 정렬)
> data[order(data$Type,data$Molecular.Weight),]복식(타입, 분자량)
> data[do.call(order,as.list(data)),]이것도 정렬하는거라는데 뭔지는 모름. (사실 데이터가 방대해서 확인 못했다)
> data[order(-data$Molecular.Weight),]> data[order(data$Smiles,-data$Type),]얘도 일단 - 붙이면 역방향으로 정렬된다.
sample()
위에서도 잠깐 나왔던 그 코드.
> v=11:20
> v
[1] 11 12 13 14 15 16 17 18 19 20
> v=sample(v)
> v
[1] 13 11 14 18 16 20 19 17 12 15근데 깔끔하게 만들어놓고 굳이 다시 개판치는 이유가 뭐냐... 머신러닝 학습용 만드나
> data=data[sample(1:nrow(data)),]데이터프레임은 이거 쓰면 된다.
as.character(), as.numeric(), as.factors()
각각 문자, 숫자, 팩터로 바꿔주는 것.
> v
[1] 13 11 14 18 16 20 19 17 12 15
> w=as.character(v)
> x=factor(v)이런 식으로 바꾼 다음 str()를 이용해 구조를 확인해보자.
> str(v)
int [1:10] 13 11 14 18 16 20 19 17 12 15
> str(w)
chr [1:10] "13" "11" "14" "18" "16" "20" "19" "17" "12" "15"
> str(x)
Factor w/ 10 levels "11","12","13",..: 3 1 4 8 6 10 9 7 2 5(끄덕)
문자와 팩터끼리는 상호전환이 되는데 숫자와 팩터는 바로 되는 게 아니라 문자 한 번 찍고 가야 한다.
> x
[1] 13 11 14 18 16 20 19 17 12 15
Levels: 11 12 13 14 15 16 17 18 19 20
> as.numeric(x)
[1] 3 1 4 8 6 10 9 7 2 5
# 뭐야 앞글자 돌려줘요팩터를 바로 numeric 줘 버리면 이렇게 된다. 어? 근데 as numeric이 낯이 익으시다고요? 실례지만 판다스 하셨음?
> z=unclass(x)
> z
[1] 3 1 4 8 6 10 9 7 2 5
attr(,"levels")
[1] "11" "12" "13" "14" "15" "16" "17" "18" "19" "20"참고로 as.numeric() 말고 unclass()로도 숫자형 된다.
duplicated()와 unique()
살다보면 중복값이 있을 때가 있다. 그걸 언제 다 세고 앉았겠음...
> a
[1] 4 12 2 26 13 2 15 17 16 7 25 14 4 -12 21 10 10 19 18
[20] 16근데 이게 중복이 있어? 있으면 그것도 웃길듯
> duplicated(a)
[1] FALSE FALSE FALSE FALSE FALSE TRUE FALSE FALSE FALSE FALSE FALSE FALSE
[13] TRUE FALSE FALSE FALSE TRUE FALSE FALSE TRUE왜 있는거죠
> a[duplicated(a)]
[1] 2 4 10 16
> unique(a[duplicated(a)])
[1] 2 4 10 16
# 얘는 반복 없이 걍 띄워주는 듯. python의 set같은 것사실 위에놈이나 밑에놈이나 그게 그거같아보이지만, unique()는 중복인 항목이 삼중 사중이어도 걍 하나로 뽑아준다. python의 set 같은 역할.
> unique(a)
[1] 4 12 2 26 13 15 17 16 7 25 14 -12 21 10 19 18
> a[!duplicated(a)]
[1] 4 12 2 26 13 15 17 16 7 25 14 -12 21 10 19 18중복이요? 뺄 수는 있음.
이건 사실 단식 벡터긴 한데 데이터프레임에서도 먹힌다.
> duplicated(data)
[1] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[13] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[25] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[37] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[49] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[61] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[73] FALSE FALSE FALSE FALSE FALSE FALSE씁 근데 이건 좀... 저거 개 방대함...
> duplicated(data$Type)
[1] FALSE TRUE TRUE FALSE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[13] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE FALSE TRUE FALSE TRUE
[25] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[37] TRUE TRUE TRUE TRUE TRUE FALSE TRUE TRUE TRUE TRUE TRUE TRUE
[49] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[61] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[73] TRUE TRUE TRUE TRUE TRUE TRUE그래서 일단 분자 타입을 픽해보겠다.
고유값 다 어디갔......
> data[duplicated(data),]
[1] ChEMBL.ID Name
[3] Synonyms Type
[5] Max.Phase Molecular.Weight
[7] Targets Bioactivities
[9] AlogP Polar.Surface.Area
[11] HBA HBD
[13] X.RO5.Violations X.Rotatable.Bonds
[15] Passes.Ro3 QED.Weighted
[17] CX.Acidic.pKa CX.Basic.pKa
[19] CX.LogP CX.LogD
[21] Aromatic.Rings Structure.Type
[23] Inorganic.Flag Heavy.Atoms
[25] HBA..Lipinski. HBD..Lipinski.
[27] X.RO5.Violations..Lipinski. Molecular.Weight..Monoisotopic.
[29] Molecular.Species Molecular.Formula
[31] Smiles데이터프레임 전역에 대한 결과.
저기 겹치는 항목이 왜 있지? 라고 생각하실 수도 있는데 ChEMBL빨 데이터가 생각보다 공백이 많다. 그래서 scatter plot 그릴 때도 dropna() 처리하고 그렸다. 그리고 그렇게 날려먹으면 못 쓰는거 꽤 많다. (dropna()가 특정 컬럼만 되는 게 아니라 묻따않 공백 빠이염이 되버림)
> data[!duplicated(data$Name),]이렇게 하면 특정 컬럼에서 겹치는 걸 볼 수 있다.
> unique(data$Name)
[1] IODINE I 131 DERLOTUXIMAB BIOTIN
[3] biotin-geranylpyrophosphate BIOTIN
4 Levels: BIOTIN ... IODINE I 131 DERLOTUXIMAB BIOTIN아 픽도 됩니다. (해당 DB는 바이오틴 관련 compound) 근데 compound 80몇개 중에 이름 있는 게 저거뿐인 건 좀 심한 거 아니냐고...
NA 들어간 것 비교하기
> df <- data.frame( a=c(TRUE,TRUE,TRUE,FALSE,FALSE,FALSE,NA,NA,NA),
+ b=c(TRUE,FALSE,NA,TRUE,FALSE,NA,TRUE,FALSE,NA))
> df
a b
1 TRUE TRUE
2 TRUE FALSE
3 TRUE NA
4 FALSE TRUE
5 FALSE FALSE
6 FALSE NA
7 NA TRUE
8 NA FALSE
9 NA NA이걸로 해 볼건데...
일단 NA는 결측값이라(Null은 아예 빈 거고 얘는 결측값으로 채워져 있는 상태) 비교가...
> df$a == df$b
[1] TRUE FALSE NA FALSE TRUE NA NA NA NA안된다.
파이썬 판다스는 그래서 NA 빼고 계산한다.
> data.frame(df, isSame = (df$a==df$b))
a b isSame
1 TRUE TRUE TRUE
2 TRUE FALSE FALSE
3 TRUE NA NA
4 FALSE TRUE FALSE
5 FALSE FALSE TRUE
6 FALSE NA NA
7 NA TRUE NA
8 NA FALSE NA
9 NA NA NA한 쪽이라도 NA가 있으면 비교를 거부하는 모습이다. 이걸 쿡북에서는 함수 짜서 해결 봤는데
> compareNA=function(v1,v2){
+ same=(v1==v2)|(is.na(v1)&is.na(v2))
+ same[is.na(same)]=FALSE
+ return(same)
+ }그게 이거다.
> compareNA(df$a,df$b)
[1] TRUE FALSE FALSE FALSE TRUE FALSE FALSE FALSE TRUE> data.frame(df,isSame = compareNA(df$a,df$b))
a b isSame
1 TRUE TRUE TRUE
2 TRUE FALSE FALSE
3 TRUE NA FALSE
4 FALSE TRUE FALSE
5 FALSE FALSE TRUE
6 FALSE NA FALSE
7 NA TRUE FALSE
8 NA FALSE FALSE
9 NA NA TRUE일단 NA랑 NA가 있으면 같은걸로 쳐주는 듯.
데이터 recoding하기
역시나 둘 다 plyr이 있어야 한다... 여기서는 크게 범주형 자료와 수치형 자료에 대한 리코딩을 진행할 것이다.
두 데이터간 차이는 숫자로 측정이 되느냐 안 되느냐 여부. 일단 범주형 데이터의 예시로는 분자의 타입이 있고, 수치형 데이터의 예시로는 분자량이 있다.
> data$Type
[1] Small molecule Small molecule Small molecule Small molecule
[6] Small molecule Small molecule Small molecule Small molecule
[11] Small molecule Small molecule Small molecule Small molecule Small molecule
[16] Small molecule Small molecule Small molecule
[21] Antibody Small molecule Protein Small molecule Small molecule
[26] Small molecule Small molecule Small molecule Small molecule Small molecule
[31] Small molecule Small molecule Small molecule Small molecule
[36] Small molecule Small molecule Small molecule
[41] Small molecule Unknown Small molecule Small molecule Small molecule
[46] Small molecule Small molecule Small molecule Small molecule Small molecule
[51] Small molecule Small molecule Small molecule Unknown Small molecule
[56] Small molecule Small molecule Small molecule Small molecule
[61] Small molecule Small molecule Small molecule Small molecule Small molecule
[66] Small molecule Small molecule Small molecule Small molecule Small molecule
[71] Small molecule Small molecule Small molecule Small molecule Small molecule
[76] Small molecule Small molecule Small molecule
Levels: Antibody Protein Small molecule Unknown이게 분자 타입. 작은 분자냐 항체냐 모르는거냐 단백질이냐에 따라 나눈다. 공백 뭐냐 즉, 수치로 측정할 수 있는 자료가 아니다.
> data$code=revalue(data$Type,c("Antibody"="A","Small molecule"="S","Protein"="P","Unknown"="U"))
> data$code
[1] S S S S S S S S S S S S S S S S A S P S S S S S S S S S S S S S
[39] S S U S S S S S S S S S S S U S S S S S S S S S S S S S S S S S S S S S
[77] S S
Levels: A P S Urevalue()
> data$code=mapvalues(data$Type,from=c("Antibody","Small molecule","Protein","Unknown"),to=c("A","S","P","U"))
> data$code
[1] S S S S S S S S S S S S S S S S A S P S S S S S S S S S S S S S
[39] S S U S S S S S S S S S S S U S S S S S S S S S S S S S S S S S S S S S
[77] S S
Levels: A P S Umapvalues()
> data$code[data$Type=="Antibody"]="A"아 상여자는 라이브러리따원 쓰지 않는다네 그리고 저걸 일일이 다 쳐야된다
다음 예시를 보자.
> data$Molecular.Weight
[1] 258.34 453.57 980.28 618.71 1838.31 634.78 919.07 789.01 650.05
[10] 1839.29 582.12 847.94 729.44 1646.16 641.93 718.87 814.15 602.71
[19] 814.97 814.02 NA 584.84 3269.70 1041.18 1075.19 700.86 605.75
[28] 1942.31 444.56 565.67 947.12 500.67 1625.96 913.05 542.68 923.15
[37] 1351.45 590.66 574.66 667.87 913.15 2532.95 1265.49 668.86 555.53
[46] 381.55 1187.32 566.12 682.82 651.23 1323.40 806.99 631.75 2625.32
[55] 326.43 764.39 606.62 1460.41 747.94 786.91 1074.26 429.52 786.99
[64] 1058.26 1474.77 549.67 1234.09 1278.10 244.32 854.06 891.95 718.84
[73] 773.45 728.92 834.14 923.15 1753.23 615.75바이오틴과 관련 있는 분자들의 분자량이다. NA가 좀 거슬리긴 하지만 패스. 분자량은 숫자로 측정하는 데이터 중 하나이다.
> data$category[data$Molecular.Weight > 500]="Large"
> data$category[data$Molecular.Weight <= 500]="Small"
> data$category
[1] "Small" "Small" "Large" "Large" "Large" "Large" "Large" "Large" "Large"
[10] "Large" "Large" "Large" "Large" "Large" "Large" "Large" "Large" "Large"
[19] "Large" "Large" NA "Large" "Large" "Large" "Large" "Large" "Large"
[28] "Large" "Small" "Large" "Large" "Large" "Large" "Large" "Large" "Large"
[37] "Large" "Large" "Large" "Large" "Large" "Large" "Large" "Large" "Large"
[46] "Small" "Large" "Large" "Large" "Large" "Large" "Large" "Large" "Large"
[55] "Small" "Large" "Large" "Large" "Large" "Large" "Large" "Small" "Large"
[64] "Large" "Large" "Large" "Large" "Large" "Small" "Large" "Large" "Large"
[73] "Large" "Large" "Large" "Large" "Large" "Large"얘는 또 category여...? 아, 여기서는 분자량 500을 기준으로 나누었다. 기준에 대해서는 나중에 또 알아보도록 하자.
> data$category=cut(data$Molecular.Weight, breaks=c(0,500,Inf),labels=c("Small","Large"))
> data$category
[1] Small Small Large Large Large Large Large Large Large Large Large Large
[13] Large Large Large Large Large Large Large Large <NA> Large Large Large
[25] Large Large Large Large Small Large Large Large Large Large Large Large
[37] Large Large Large Large Large Large Large Large Large Small Large Large
[49] Large Large Large Large Large Large Small Large Large Large Large Large
[61] Large Small Large Large Large Large Large Large Small Large Large Large
[73] Large Large Large Large Large Large
Levels: Small Large저렇게 여러 줄 치기 귀찮으면 cut()으로 나누면 된다.
컬럼간 연산
> data$ratio=data$HBA/data$HBD
경고메시지(들):
In Ops.factor(data$HBA, data$HBD) :
요인(factors)에 대하여 의미있는 ‘/’가 아닙니다.
> data$ratio
[1] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[26] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[51] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[76] NA NA NA근데 저거 엄연히 수치가 있는건데 왜 NA가 뜨는겨...
> data$HBA
[1] 4 7 12 10 None 9 17 13 12 None 9 14 8 None 10
[16] 10 9 9 14 12 8 None None None 11 7 None 7 9
[31] 17 7 None 19 5 15 None 10 9 7 16 None None 9 7
[46] 6 None 8 10 9 None 8 11 None 4 10 7 None 10 14
[61] None 4 13 None None 8 None None 3 11 15 10 9 10 11
[76] 15 None 10
Levels: 10 11 12 13 14 15 16 17 19 3 4 5 6 7 8 9 None
> data$HBD
[1] 2 5 6 3 None 8 3 3 6 None 6 12 3 None 8
[16] 8 5 2 3 9 3 None None None 8 3 None 3 6
[31] 3 3 None 13 5 3 None 3 2 5 4 None None 7 6
[46] 3 None 5 5 8 None 8 6 None 3 9 6 None 9 3
[61] None 4 3 None None 5 None None 3 5 13 6 3 6 5
[76] 3 None 5
Levels: 12 13 2 3 4 5 6 7 8 9 None내가 있다고 했잖음.
> data$sum = as.numeric(data$HBA) + as.numeric(data$HBD)
> data$sum
[1] 16 22 12 7 30 27 14 10 12 30 25 8 21 30 12 12 24 21 11 15 2 21 30 30 30
[26] 13 20 30 20 25 14 20 30 13 20 12 30 7 21 22 14 30 30 26 23 19 30 23 9 27
[51] 30 26 11 30 17 13 23 30 13 11 30 18 10 30 30 23 30 30 16 10 10 10 22 10 10
[76] 12 30 9(마른세수) 쟤네 팩터라 숫자로 바로 바꾸면 안된다.
> data$sum = as.numeric(as.character(data$HBA)) + as.numeric(as.character(data$HBD))
경고메시지(들):
1: 강제형변환에 의해 생성된 NA 입니다
2: 강제형변환에 의해 생성된 NA 입니다
> data$sum
[1] 6 12 18 13 NA 17 20 16 18 NA 15 26 11 NA 18 18 14 11 17 21 NA 11 NA NA NA
[26] 19 10 NA 10 15 20 10 NA 32 10 18 NA 13 11 12 20 NA NA 16 13 9 NA 13 15 17
[51] NA 16 17 NA 7 19 13 NA 19 17 NA 8 16 NA NA 13 NA NA 6 16 28 16 12 16 16
[76] 18 NA 15이중변환 해야 한다...
벡터 매핑하기
여기서도 plyr이 쓰일 줄은 몰랐고...
> str=c("alpha","omicron","pi")
> revalue(str,c("alpha"="sigma"))
[1] "sigma" "omicron" "pi"> mapvalues(str,from=c("alpha"),to=c("sigma"))
[1] "sigma" "omicron" "pi"벡터값 매핑도 revalue()와 mapvalues()로 한다. 단, revalues()는 숫자 매핑은 안된다.
> mapvalues(v,from=c(1,2),to=c(11,12))
[1] 3 6 8 9 12 5 7 4 10 11근데 시그마 하니 록맨 X의 시그마가 생각나는군... X5 스테이지 브금이 아주 클럽 브금이었음...
생각난 김에 듣고 가자. 아니 이게 무슨 흐름이지
> str[str=="alpha"]="sigma"
> str
[1] "sigma" "omicron" "pi"> v[v==2]=12
> v
[1] 3 6 8 9 12 5 7 4 10 1라이브러리 없이도 되긴 된다. 값이 많으면 좀 귀찮아지긴 하겠지만...
> sub("^sigma$","omicron",str)
[1] "omicron" "omicron" "pi"
> gsub("o","O",str)
[1] "sigma" "OmicrOn" "pi"sub()과 gsub()도 된다고 합니다.
'Coding > R' 카테고리의 다른 글
| R 배워보기- 6.3. Manipulating data-Data Frames (0) | 2022.08.20 |
|---|---|
| R 배워보기- 6.2. Manipulating data-Factors (0) | 2022.08.20 |
| R 배워보기-5. 데이터 불러오고 쓰기 (0) | 2022.08.20 |
| 번외편-R로 미적분 하기 (0) | 2022.08.20 |
| R 배워보기-4. 공식 (0) | 2022.08.20 |
MSA: multiple sequence alignment
여기에 관한 이론적인 설명은 나중에 또 입 털어드림 ㅇㅇ
아 참고로 MSA 관련해서 다른건 다 결과가 제대로 나왔는데 툴 관련해서 결과가 안나왔어요
이게 암만 찾아도 답이 없어서 좀 더 알아보고 수정할 가능성이 있음
시범조교 앞으로
오늘은 시범조교 가짓수가 좀 많은데 그 중에서도 FASTA 파일들을 좀 올리고자 한다. 이거 말고도 pfam에서 두갠가 받았는데 그건 걍 가서 암거나 받으면 된다.
해당 파일은 박테리아의 16s rRNA 시퀀스가 들어있는 파일이다. FASTA 파일이라 메모장 있으면 일단 열 수는 있다. 리눅스에서는 gedit으로 만들고 편집하고 다 했다. (vim 안씀) rRNA 시퀀스는 Silva에서 가져왔다. 고마워요 실바!
Agrobacterium
A. radiobacter를 필두로 하는 뿌리혹세균들(밑에 두놈도 뿌리에 혹 만드는지는 모름)
-Agrobacterium
Agrobacterium radiobacter (구 Agrobacterium tumefaciens)
Agrobacterium agile
Agrobacterium pusense
Agrobacterium salinitolerans
-Rhizobium
Rhizobium tropici (일반적으로 알고 있는 뿌리혹박테리아)
Rhizobium hainanense
Rhizobium gallicum
Rhizobium fabae
-Hoeflea
Hoeflea alexandrii
Hoeflea halophila 웬지 짠 거 좋아하게 생겼는데? 할로박테리움같은건가
Hoeflea trophica
-Ciceribacter
Ciceribacter lividus
Ciceribacter azotifigens
Ciceribacter thiooxidans
Enterobacter
E.coli를 필두로 하는 장내 세균들
-Enterococcus
Enterococcus faecalis (어디서 많이 봤음)
Enterococcus hirae
Enterococcus avium
Enterococcus caccae
-Escherichia
Escherichia coli (그냥 어디서나 볼 수 있는 대장균)
Escherichia coli O157:H7 (감염되면 X되는 대장균)
Escherichia albertii
Escherichia fergusonii
-Shigella
Shigella boydii
Shigella sonnei
Shigella dysenteriae
Shigella flexneri
Lactobacillus
님들 많이 드시는 그 유산균 맞습니다.
-Lactobacillus
Lactobacillus acidophilus
Lactobacillus helveticus
Lactobacillus plantarum (이분도 김치에서 발견된다)
Lactobacillus thailandensis DSM 22698 = JCM 13996 (아마 뒤에껀 strain 이름인 듯)
-Leuconostoc
Leuconostoc carnosum
Leuconostoc mesenteroides
Leuconostoc kimchii (이건 있다)
Leuconostoc miyukkimchii (저자 나와봐요 미역김치는 무슨 저세상 학명이야)
-Bifidobacterium (얘네는 방선균인데 일단 유산균으로 섭취 하기는 함... 비피더스 뭐 이런거)
Bifidobacterium bifidum
Bifidobacterium actinocoloniiforme DSM 22766 (아마 strain 이름...)
Bifidobacterium catenulatum
아니 근데 미역김치 학명 실화냐 대체 어디서 발견하면 학명이 저렇게 되는건데요 아니 저기 김치 하나 있는거 뭔데 김치요 참고로 C.kimchii(구 L.kimchii)는 데이터 없어서 못 넣었음... 김치 파티 각 나왔다
시퀀스 가져오기
MSA를 할래도 시퀀스를 가져와야 한다. 그것도 하나 말고 뭉텅이로. 여기서도 read()와 parse()로 나뉘긴 한데, 이거는 기존에 SeqIO로 읽을 때처럼 시퀀스 갯수가 여러 개 있어도 single alignment면 read()로 읽을 수 있다.
참고로 쿡북 예제로 스톡홀름 파일이 나오긴 했는데 FASTA도 불러올수는 있다.
from Bio import AlignIO
alignment = AlignIO.read("/home/koreanraichu/agrobacterium.fasta", "fasta")
print(alignment)MSA를 할 때는 SeqIO가 아니라 AlignIO로 불러오면 된다.
Alignment with 14 rows and 1561 columns
AUUCUCAACUUGAGAGUUUGAUCCUGGCUCAGAACGAACGCUGG...AAG AB102732.157.1651
................................AACGAACGCUGG...... AB680818.1.1406
................................AUUGAACGCUGG...... AB680959.1.1462
................................AACGAACGCUGG...... AB682466.1.1406
................................AACGAACGCUGG...... AB682468.1.1406
......................................CGCUGG...... AB969785.1.1417
............AGAGUUUGAUCCUGGCUCAGAACGAACGCUGG...... AJ786600.1.1449
.................................................. DQ835303.1.1353
.................................................. GU564401.1.1419
................................AACGAACGCUGG...... JF957616.1.1410
.................................................. JQ230000.1.1392
.................................................. KP142169.1.1341
............AGAGUUUGAUCAUGGCUCAGAACGAACGCUGG...... KU975391.1.1451
...............................A-ACGAACGCUGG...... KX510117.1.1407여기서 말하는 컬럼은 글자 하나. PDB파일처럼 얘도 알파벳 하나가 컬럼이다.
for record in alignment:
print(record.id, record.description) # 예제 코드는 Sequence와 ID를 출력하라고 했는데 기니까 다른걸로 바꿔보자.AB102732.157.1651 AB102732.157.1651 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Agrobacterium radiobacter
AB680818.1.1406 AB680818.1.1406 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Rhizobium tropici
AB680959.1.1462 AB680959.1.1462 Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Pseudomonadaceae;Pseudomonas;Agrobacterium agile
AB682466.1.1406 AB682466.1.1406 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Rhizobium hainanense
AB682468.1.1406 AB682468.1.1406 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Rhizobium gallicum
AB969785.1.1417 AB969785.1.1417 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Agrobacterium pusense
AJ786600.1.1449 AJ786600.1.1449 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Hoeflea;Hoeflea alexandrii
DQ835303.1.1353 DQ835303.1.1353 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Rhizobium fabae
GU564401.1.1419 GU564401.1.1419 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Hoeflea;Hoeflea halophila
JF957616.1.1410 JF957616.1.1410 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Hoeflea;Hoeflea phototrophica
JQ230000.1.1392 JQ230000.1.1392 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Ciceribacter;Ciceribacter lividus
KP142169.1.1341 KP142169.1.1341 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Agrobacterium salinitolerans
KU975391.1.1451 KU975391.1.1451 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Ciceribacter;Ciceribacter thiooxidans
KX510117.1.1407 KX510117.1.1407 Bacteria;Proteobacteria;Alphaproteobacteria;Rhizobiales;Rhizobiaceae;Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium;Ciceribacter azotifigens이런 식으로 레코드 만들어도 된다. 예제에는 ID랑 시퀀스로 되어 있던거 ID랑 description으로 바꿨다... 시퀀스가 너무 길어... 출력 format을 지정하거나 쌩으로 레코드를 가져올 수도 있는데, 길이가... 길이가...... 정말 장난없다...
from Bio import AlignIO
alignments = AlignIO.parse("/home/koreanraichu/lactobacillus.fasta", "fasta")
for alignment in alignments:
print(alignment)Alignment with 12 rows and 1606 columns
...............UUUGAUCAUGGCUCAGGACGAACGCUGGC...... AB008203.1.1553
................UUGAUCAUGGCUCAGGACGAACGCUGGC...... AB008210.1.1552
.........................................CGC...... AB022925.1.1450
..........................................GC...... AB023242.1.1446
............GAGUUUGAUCCUGGCUCAGGACGAACGCUGGC...... AB112083.1.1557
...........GGGUUUCGAUUCUGGCUCAGGAUGAACGCUGGC...... AB437354.1.1520
.............GUUUCGAUUCUGGCUCAGGAUGAACGCUGGC...... AB437356.1.1519
...............................GAUGAACGCUGGC...... AF173986.1.1505
...........AGAGUUUGAUCCUGGCUCAGGAUGAACGCUGGC...... AYZK01000017.137.1688
UUUUUUUGUGGAGGGUUUGAUUCUGGCUCAGGAUGAACGCUGGC...AGA CP011786.1420993.1422533
UUUUUUUGUGGAGGGUUUGAUUCUGGCUCAGGAUGAACGCUGGC...AGA CP011786.1514900.1516440
...............................GAUGAACGCUGGC...... KX232108.1.1480Parse로 가져와도 똑같긴 한데 SeqIO나 얘나 파싱으로 가져오면 for문 있어야 제대로 뜬다.
from Bio import AlignIO
handle="/home/koreanraichu/lactobacillus.fasta"
for alignments in AlignIO.parse(handle, "fasta",seq_count=2):
print("Alignment lentgh %i" % alignments.get_alignment_length())
for record in alignments:
print(record.description)Alignment lentgh 1606
AB008203.1.1553 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lactobacillus;Lactobacillus acidophilus
AB008210.1.1552 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lactobacillus;Lactobacillus helveticus
Alignment lentgh 1606
AB022925.1.1450 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Leuconostoc;Leuconostoc carnosum
AB023242.1.1446 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Leuconostoc;Leuconostoc mesenteroides
Alignment lentgh 1606
AB112083.1.1557 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lactiplantibacillus;Lactobacillus plantarum
AB437354.1.1520 Bacteria;Actinobacteriota;Actinobacteria;Bifidobacteriales;Bifidobacteriaceae;Bifidobacterium;Bifidobacterium adolescentis
Alignment lentgh 1606
AB437356.1.1519 Bacteria;Actinobacteriota;Actinobacteria;Bifidobacteriales;Bifidobacteriaceae;Bifidobacterium;Bifidobacterium bifidum
AF173986.1.1505 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Leuconostoc;Leuconostoc kimchii
Alignment lentgh 1606
AYZK01000017.137.1688 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lacticaseibacillus;Lactobacillus thailandensis DSM 22698 = JCM 13996
CP011786.1420993.1422533 Bacteria;Actinobacteriota;Actinobacteria;Bifidobacteriales;Bifidobacteriaceae;Bifidobacterium;Bifidobacterium actinocoloniiforme DSM 22766
Alignment lentgh 1606
CP011786.1514900.1516440 Bacteria;Actinobacteriota;Actinobacteria;Bifidobacteriales;Bifidobacteriaceae;Bifidobacterium;Bifidobacterium actinocoloniiforme DSM 22766
KX232108.1.1480 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Leuconostoc;Leuconostoc miyukkimchii끊어뽑기도 된다. 코드에 있는 숫자 수정하면 3개 4개 묶는 것도 된다.
쓰기
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio.Align import MultipleSeqAlignment
from Bio import AlignIO
align1=MultipleSeqAlignment([
SeqRecord(Seq("GGCC"),id="HaeIII"),
SeqRecord(Seq("G-CC"),id="id2"),
SeqRecord(Seq("G--C"),id="id3")
])
align2=MultipleSeqAlignment([
SeqRecord(Seq("GAATTC"),id="EcoRi"),
SeqRecord(Seq("G-ATTC"),id="id5"),
SeqRecord(Seq("G--TTC"),id="id6")
])
my_alignment=[align1,align2]
# 여기까지 시퀀스를 만들고
AlignIO.write(my_alignment, "/home/koreanraichu/sequence.fasta", "fasta")
# 여기서 저(별)장나 저거 IL-16 전에 썼던거 안된 이유 알아냈음... ㅋㅋㅋㅋㅋㅋ Seq=("")가 아니라 Seq("")였다. 아무튼 이런 식으로 쓸 수 있고 주석 있어서 대충 아시겠지만 순서대로 Alignment 요소를 만들고->시퀀스 만들고->파일로 저장하는 코드이다.
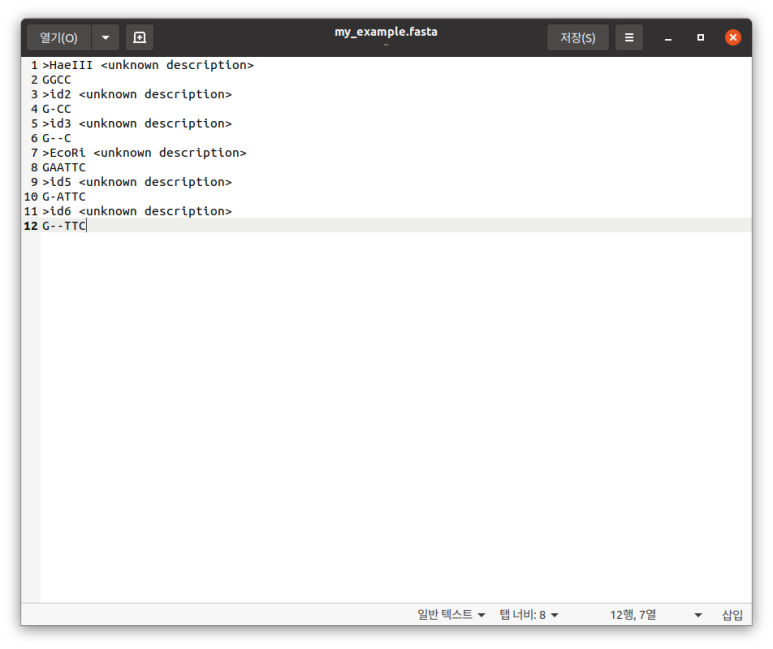
이런 식으로 저장된다. (그야 내가 FASTA로 저장했으니까...)
참고로 미리 말해두겠지만 본인 리눅스에서 FASTA, PHYLIP, 스톡홀름, clustalW 다 지에딧으로 연다. 그렇게 열리고…
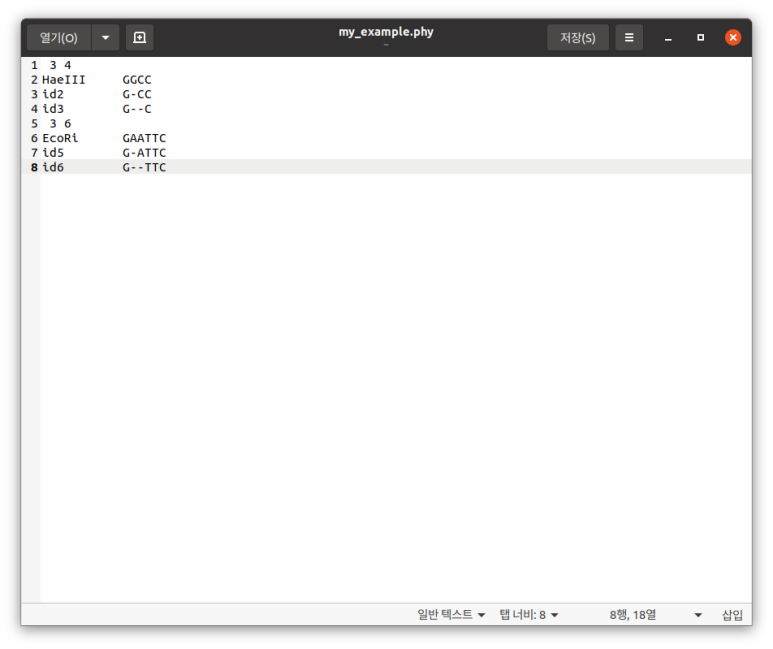
이게 필립 파일인데 얘는 변환하려면 시퀀스 길이가 다 같아야 하고, 공백이 -이어야 한다. 공백에 . 있으면 지원 안 한다고 오류 토한다.
선생님 이거 변환 됩니까?
변환이요? 뭐 클러스탈이나 스톡뭐시기 말하는거면 된다.
count = AlignIO.convert("/home/koreanraichu/agrobacterium.fasta", "fasta", "/home/koreanraichu/agrobacterium.sth",
"stockholm")
print("Converted %i alignments" % count)alignments = AlignIO.parse("/home/koreanraichu/agrobacterium.fasta", "fasta")
count = AlignIO.write(alignments, "/home/koreanraichu/agrobacterium.aln","clustal")
print("Converted %i alignments" % count)
# ClustalW위는 convert를 사용해 스톡홀름 파일로 바꾼거고, 아래는 parse와 write를 사용해 클러스탈로 바꾼 것이다.
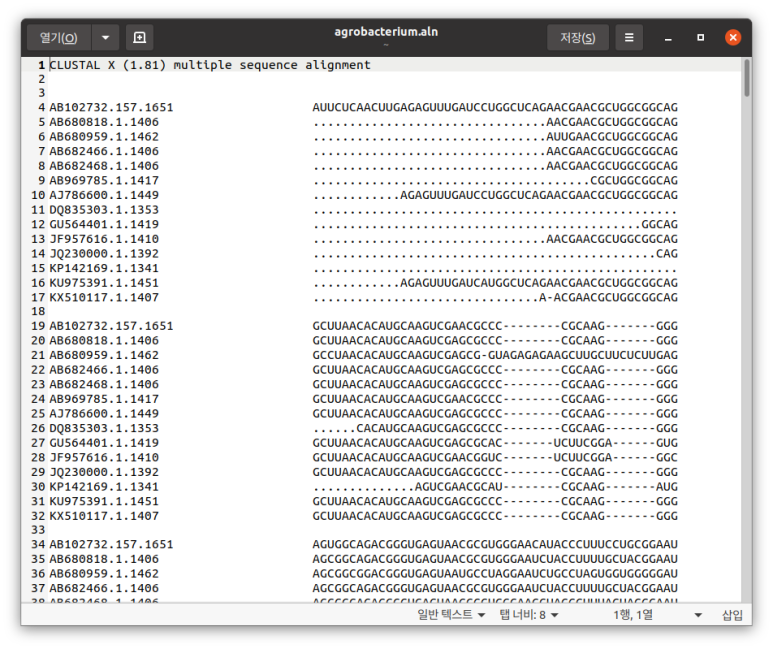
위가 clustalw, 아래가 스톡홀름 포맷.
alignment = AlignIO.read("/home/koreanraichu/PF00096_seed.txt", "stockholm")
AlignIO.write([alignment], "PF05371_seed.aln", "clustal")
print("Converted %i alignments" % count)
# read 후 리스트화해서 변환read로 읽은 다음 리스트화해서 변환하는 것도 된다.
count = AlignIO.convert("/home/koreanraichu/PF08449_seed.txt", "stockholm", "/home/koreanraichu/PF08449_seed.phy",
"phylip")
print("Converted %i alignments" % count)
# 이거라면 필립 될거같은데?공백이 -이고 시퀀스 bp가 전부 동일하다는 전제하에 Phylip 포맷으로 만드는 것도 된다.
alignment2 = AlignIO.read("/home/koreanraichu/PF08449_seed.txt", "stockholm")
name_mapping = {}
for i, record in enumerate(alignment):
name_mapping[i] = record.id
record.id = "seq%i" % i
AlignIO.write([alignment], "PF08449_seed_ID.phy", "phylip")
# 오 뭔진 모르겠지만 ID가 숫자가 된 건가딕셔너리화 한 다음 아이디 새로 지정해주는 것도 된다. 위 코드를 실행하면
이렇게 된다. 잘 보면 ID 영역에 seq(숫자)가 들어가 있다.
출력 형식만 바꾸기
이건 뭔 소리냐... 여는 파일은 FASTA파일인데 파일 수정 없이 clustalw나 스톡홀름 형식으로 출력할 수 있다. (phylip은 아마 글자수 같고 공백 -여야 먹힐듯) 참고로 결과는 길어서 생략.
from Bio import AlignIO
alignment = AlignIO.read("/home/koreanraichu/lactobacillus.fasta", "fasta")
print(format(alignment, "clustal"))해당 코드는 FASTA파일을 클러스탈 형식으로 보여달라는 얘기고, 스톡홀름으로 볼 수도 있다.
슬라이싱
넘파이 배열 자르는것처럼 얘도 슬라이싱이 된다. (넘파이 2차원 배열 잘라먹는 거 생각하면 된다)
from Bio import AlignIO
alignment = AlignIO.read("/home/koreanraichu/PF08449_seed.txt", "stockholm")
print(alignment[0:5])ISLIPISMIMVGCCSNVISLELIMKQSQ-SH-A---------IL...TSG S35B4_DICDI/7-328
SFVLILSLVFGGCCSNVISFEHMVQGSN-INLG---------NI...YGS YEA4_KLULA/2-321
NSLKAFALVFGGCCSNVITFETLMSNET-GSIN---------NL...LGS YEA4_YEAST/3-327
MIASALSFIFGGCCSNAYALEALVREFP-SS-G---------IL...SAR YEA4_SCHPO/1-304
--GVMLSLIFGGCCSNVFALESIIKVEP-GA-G---------TL...L-- Q1K947_NEUCR/81-406이렇게 하면 pandas의 head()처럼 위에서 다섯개만 보여준다.
print(alignment[0,1]) # 0번째 인덱스의 두번째 컬럼
S이런 식으로 2차원 인덱싱과 슬라이싱도 되고
print(alignment[:,6]) # 무슨 컬럼을 가져온거냐
SSASSGTCLSSASLASL이거는 전체 인덱스에서 일곱번째 글자들을 가져온 것. (실화다)
print(alignment[0:5,0:5]) # index+column 범위를 지정할 수 있다
ISLIP S35B4_DICDI/7-328
SFVLI YEA4_KLULA/2-321
NSLKA YEA4_YEAST/3-327
MIASA YEA4_SCHPO/1-304
--GVM Q1K947_NEUCR/81-406이런 식으로 범위 지정해서 잘라오는 것도 되고 특정 인덱스, 컬럼부터 다 가져오는 것도 된다. 저거 저대로 잘라먹는 것도 되고, Numpy 데려와서 배열화하는 것도 가능하다. 근데 예제대로 했더니 오류남... (안되는 건 아닌데 오류뜬다)
본격적인 정보 얻기
아까까지 대체 뭐 한겨...
from Bio import AlignIO
alignment = AlignIO.read("/home/koreanraichu/lactobacillus.fasta", "fasta") # 아이고 유산균씨 오랜만입니다
substitutions = alignment.substitutions
print(substitutions) . A C G U
. 1365.0 321.0 310.5 461.5 406.0
A 321.0 19985.0 791.0 2218.5 978.5
C 310.5 791.0 18856.0 957.5 2048.0
G 461.5 2218.5 957.5 26642.0 1015.0
U 406.0 978.5 2048.0 1015.0 16007.0이거는 rRNA라 티민 대신 우라실이 나왔다. 저건 아마도 해당 염기가 다른걸로 바뀐 비율? 횟수? 이런 걸 봐주는 것 같다. 예를 들자면 아데닌이 시토신, 구아닌, 우라실로 바뀌는 뭐 그런거. (공백 공백은 뭐여...) 저기서 옵션으로 .select() 주면 출력 순서가 바뀐다. (예: .select(“AUGC.”)으로 주면 행렬 인덱스랑 컬럼 순서가 AUCG.로 바뀐다)
Tool
이 부분이 문제가 많아요... OTL 둘 다 깔고 해야 하나...
ClustalW
from Bio.Align.Applications import ClustalwCommandline
cline = ClustalwCommandline("clustalw2", infile="/home/koreanraichu/lactobacillus.fasta")
print(cline)이런 식으로 불러서
clustalw2 -infile=/home/koreanraichu/lactobacillus.fasta니가 왜 거기서 나옴???
이거 뭐 트리 만드는 파일 만들어준다매 왜 안해줘요. 이럴거면 그냥 메가 쓰면 안되냐
MUSCLE
from Bio.Align.Applications import MuscleCommandline
cline = MuscleCommandline(input="/home/koreanraichu/lactobacillus.aln", out="/home/koreanraichu/lactobacillus.txt")
print(cline)얘는 이런 식으로 부르면
muscle -in /home/koreanraichu/lactobacillus.aln -out /home/koreanraichu/lactobacillus.txt이러시는 이유가 있으실 것 아니예요.
from Bio.Align.Applications import MuscleCommandline
cline = MuscleCommandline(input="/home/koreanraichu/lactobacillus.fasta", out="/home/koreanraichu/lactobacillus.txt",
clw=True)
print(cline)
muscle -in /home/koreanraichu/lactobacillus.fasta -out /home/koreanraichu/lactobacillus.txt -clw이걸 쓰면 clustalW 비슷하게 출력된다. (물론 파일은 생기지 않았습니다)
from Bio.Align.Applications import MuscleCommandline
cline = MuscleCommandline(input="/home/koreanraichu/lactobacillus.fasta", out="/home/koreanraichu/lactobacillus.txt",
clwstrict=True)
print(cline)
muscle -in /home/koreanraichu/lactobacillus.fasta -out /home/koreanraichu/lactobacillus.txt -clwstrict이걸 쓰면 clustalW headline을 쓴다는데 파일이 없다. 착한 사람만 보이는 파일인가... 그래서 얘네는 못했다.
Pairwise
왜 블래스트에서 뭐 검색하면 결과에 줄 그어서 나오는 그거 말하는 거 맞다.
from Bio import pairwise2
from Bio import SeqIO
seq1 = SeqIO.read("/home/koreanraichu/alpha.faa", "fasta")
seq2 = SeqIO.read("/home/koreanraichu/beta.faa", "fasta")
alignments = pairwise2.align.globalxx(seq1.seq, seq2.seq)
print(alignments)근데 이거 파싱으로 개별 레코드끼리 비교 못하나... 나중에 다시 함 트라이해 볼 예정.
[Alignment(seqA='MV-LSPADKTNV---K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----T--PEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=217), Alignment(seqA='MV-LSPADKTNV--K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----T-PEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LSPADKTNV--K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----TP-EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LS-PADKTNV--K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-TP------EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=217), Alignment(seqA='MV-LSPADKTNV--K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHLTP------EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LSPADKTN-V-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----T-PEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LSPADKTNV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----TPEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=215), Alignment(seqA='MV-LS-PADKTNV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-TP-----EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LSPADKTNV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHLTP-----EEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=215), Alignment(seqA='MV-LSPADKT-NV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-', seqB='MVHL-----TPE-EKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H', score=72.0, start=0, end=216), Alignment(seqA='MV-LS-PADKTNV-K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-뭔진 모르겠지만 제가 잘못했습니다. (저거 len으로 보면 길이 장난없다)
print(pairwise2.format_alignment(*alignments[0]))이렇게 쳐 주면 비로소 우리가 생각하는(?)
MV-LSPADKTNV---K-A--A-WGKVGAHAG---EY-GA-EALE-RMFLSF----PTTK-TY--FPHFDL-SH-G--S---AQVK-G------HGKKV--A--DA-LTNAVAHVD-DMPNALS----A-LSD-LHAH--KLR-VDPV-NFKL-LSHCL---LVT--LAAHLPA----EFTPA-VH-ASLDKFLAS---VSTV------LTS--KYR-
|| | | | | | |||| | | ||| | | | | | | | | | | | | ||||| | | | || | | | | || | | || ||| || | | || | || |||| | | | | | | ||
MVHL-----T--PEEKSAVTALWGKV-----NVDE-VG-GEAL-GR--L--LVVYP---WT-QRF--F--ES-FGDLSTPDA-V-MGNPKVKAHGKKVLGAFSD-GL----AH--LD--N-L-KGTFATLS-EL--HCDKL-HVDP-ENF--RL---LGNVLV-CVL-AH---HFGKEFTP-PV-QA------A-YQKV--VAGVANAL--AHKY-H
Score=72이 결과가 나온다. BLAST에서도 이런 식으로 나온다.
BLOSUM62와 함께 정렬...아니고 줄을
from Bio import pairwise2
from Bio import SeqIO
from Bio.Align import substitution_matrices
blosum62 = substitution_matrices.load("BLOSUM62")
seq1 = SeqIO.read("/home/koreanraichu/alpha.faa", "fasta")
seq2 = SeqIO.read("/home/koreanraichu/beta.faa", "fasta")
alignments = pairwise2.align.globalds(seq1.seq, seq2.seq,blosum62, -10, -0.5)
print(pairwise2.format_alignment(*alignments[0]))이거 [0] 빼면요? 뭔진 모르겠지만 잘못했어요 소리가 절로 나온다. (길이 장난없음)
MV-LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS-----HGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR
|| |.|..|..|.|.|||| ...|.|.|||.|.....|.|...|..| ||| .|...||.|||||..|.....||.|........||.||..||.|||.||.||...|...||.|...||||.|.|...|..|.|...|..||.
MVHLTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH
Score=292.5.globalds는 전역(전체 시퀀스)를 보는거고 .localds로 부분만 볼 수도 있다.
alignments = pairwise2.align.localds("LSPADKTNVKAA", "PEEKSAV", blosum62, -10, -1)3 PADKTNV
|..|..|
1 PEEKSAV
Score=16이런 식으로 부분부분 볼 수도 있다. 엥? 근데 저렇게 해 놓으면 어딘지 어떻게 알아요?
print(pairwise2.format_alignment(*alignments[0],full_sequences=True))LSPADKTNVKAA
|..|..|
--PEEKSAV---개발자는 다 계획이 있었다.
Pairwise aligner
from Bio import Align
aligner = Align.PairwiseAligner()
seq1="GAATTC"
seq2="GCATTC"
score = aligner.score(seq1, seq2)
print(score)
5.0일단 모셔보래서 모셔봤습니다. 이거는 score 뽑는거고
from Bio import Align
aligner = Align.PairwiseAligner() # 그래서 모셔왔습니다
seq1="GAATTC"
seq2="GCATTC"
alignments = aligner.align(seq1, seq2)
for alignment in alignments:
print(alignment)이렇게 치면
G-AATTC
|-|-|||
GCA-TTC
GA-ATTC
|--||||
G-CATTC
G-AATTC
|--||||
GC-ATTC
GAATTC
|.||||
GCATTC이렇게 나온다. 전 맨 밑에껄로 보여주세요. 픽 안되나
GAATTC
||||||
GAATTC100% 일치하는 시퀀스는 이런 식으로 보여준다. aligner.mode="local"을 주면 위에것처럼 중구난방으로 안 뽑아주고
GAATTC
|||||
CAATTC이런 식으로 뽑아준다.
Wildcard
일반적으로 검색할 때(구글이나 파일같은 거) *이 와일드카드지만, 여기서는 와일드카드를 지정할 수 있다.
# wildcard를 지정한 다음 쓸 수도 있다
aligner.wildcard = "?"
seq3="?GCC"
seq4="GGCC"
alignments2 = aligner.align(seq3, seq4)
for alignment in alignments2:
print(alignment)?G-CC
|-||
GGCC
?GCC
|||
GGCC여기서는 일단 ?로 지정해보자.
'Coding > Python' 카테고리의 다른 글
| Biopython으로 BLAST 돌려보기 (0) | 2022.08.20 |
|---|---|
| 번외편-코딩테스트 풀이 (0) | 2022.08.20 |
| Biopython SeqIO 써보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 레코드 생성하고 만져보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 다뤄보기 (0) | 2022.08.20 |
파이참 쓰다보니 키보드 먹통돼서 재부팅 여러번 했다. 이것도 갈 때 된 듯...
*참고로 그냥 리드나 파싱으로 긁어오는 방법은 첫 글에서 썼으므로 생략한다.
Iteration 활용하기
근데 들어도 이건 뭔 기능인가 싶긴 함.
from Bio import SeqIO
identifiers = [seq_record.id for seq_record in SeqIO.parse("/home/koreanraichu/sequence.gb", "genbank")]
print(identifiers)뭐야 wrap 넣어줘요...
레코드와 for문을 조합해서 원하는 데이터(ID나 시퀀스, description)만 가져올 수 있다.
from Bio import SeqIO
identifiers = [seq_record.id for seq_record in SeqIO.parse("/home/koreanraichu/ls_orchid.gbk", "genbank")]
print(identifiers)['Z78533.1', 'Z78532.1', 'Z78531.1', 'Z78530.1', 'Z78529.1', 'Z78527.1', 'Z78526.1', 'Z78525.1', 'Z78524.1', 'Z78523.1', 'Z78522.1', 'Z78521.1', 'Z78520.1', 'Z78519.1', 'Z78518.1', 'Z78517.1', 'Z78516.1', 'Z78515.1', 'Z78514.1', 'Z78513.1', 'Z78512.1', 'Z78511.1', 'Z78510.1', 'Z78509.1', 'Z78508.1', 'Z78507.1', 'Z78506.1', 'Z78505.1', 'Z78504.1', 'Z78503.1', 'Z78502.1', 'Z78501.1', 'Z78500.1', 'Z78499.1', 'Z78498.1', 'Z78497.1', 'Z78496.1', 'Z78495.1', 'Z78494.1', 'Z78493.1', 'Z78492.1', 'Z78491.1', 'Z78490.1', 'Z78489.1', 'Z78488.1', 'Z78487.1', 'Z78486.1', 'Z78485.1', 'Z78484.1', 'Z78483.1', 'Z78482.1', 'Z78481.1', 'Z78480.1', 'Z78479.1', 'Z78478.1', 'Z78477.1', 'Z78476.1', 'Z78475.1', 'Z78474.1', 'Z78473.1', 'Z78472.1', 'Z78471.1', 'Z78470.1', 'Z78469.1', 'Z78468.1', 'Z78467.1', 'Z78466.1', 'Z78465.1', 'Z78464.1', 'Z78463.1', 'Z78462.1', 'Z78461.1', 'Z78460.1', 'Z78459.1', 'Z78458.1', 'Z78457.1', 'Z78456.1', 'Z78455.1', 'Z78454.1', 'Z78453.1', 'Z78452.1', 'Z78451.1', 'Z78450.1', 'Z78449.1', 'Z78448.1', 'Z78447.1', 'Z78446.1', 'Z78445.1', 'Z78444.1', 'Z78443.1', 'Z78442.1', 'Z78441.1', 'Z78440.1', 'Z78439.1']예제에 있단 파일로 돌렸더니 94개 나온다. 근데 94개가 저렇게 중구난방으로 나오니까 힘들다... 이거 넘파이 배열로 바꿔서 깔쌈하게 보여주면 안되나?
from Bio import SeqIO
import numpy as np
identifiers = [seq_record.id for seq_record in SeqIO.parse("/home/koreanraichu/ls_orchid.gbk", "genbank")]
identifiers=np.array(identifiers)
identifiers=identifiers.reshape(2,47)
print(identifiers)그래서 해봤다.
[['Z78533.1' 'Z78532.1' 'Z78531.1' 'Z78530.1' 'Z78529.1' 'Z78527.1'
'Z78526.1' 'Z78525.1' 'Z78524.1' 'Z78523.1' 'Z78522.1' 'Z78521.1'
'Z78520.1' 'Z78519.1' 'Z78518.1' 'Z78517.1' 'Z78516.1' 'Z78515.1'
'Z78514.1' 'Z78513.1' 'Z78512.1' 'Z78511.1' 'Z78510.1' 'Z78509.1'
'Z78508.1' 'Z78507.1' 'Z78506.1' 'Z78505.1' 'Z78504.1' 'Z78503.1'
'Z78502.1' 'Z78501.1' 'Z78500.1' 'Z78499.1' 'Z78498.1' 'Z78497.1'
'Z78496.1' 'Z78495.1' 'Z78494.1' 'Z78493.1' 'Z78492.1' 'Z78491.1'
'Z78490.1' 'Z78489.1' 'Z78488.1' 'Z78487.1' 'Z78486.1']
['Z78485.1' 'Z78484.1' 'Z78483.1' 'Z78482.1' 'Z78481.1' 'Z78480.1'
'Z78479.1' 'Z78478.1' 'Z78477.1' 'Z78476.1' 'Z78475.1' 'Z78474.1'
'Z78473.1' 'Z78472.1' 'Z78471.1' 'Z78470.1' 'Z78469.1' 'Z78468.1'
'Z78467.1' 'Z78466.1' 'Z78465.1' 'Z78464.1' 'Z78463.1' 'Z78462.1'
'Z78461.1' 'Z78460.1' 'Z78459.1' 'Z78458.1' 'Z78457.1' 'Z78456.1'
'Z78455.1' 'Z78454.1' 'Z78453.1' 'Z78452.1' 'Z78451.1' 'Z78450.1'
'Z78449.1' 'Z78448.1' 'Z78447.1' 'Z78446.1' 'Z78445.1' 'Z78444.1'
'Z78443.1' 'Z78442.1' 'Z78441.1' 'Z78440.1' 'Z78439.1']]참고로 numpy array는 5*6을 만들려면 진짜 데이터가 30개 있어야된다. 즉, 저게 최선이다.
레코드 반복
이것도 Iteration을 이용한다.
from Bio import SeqIO
record_iterator = SeqIO.parse("/home/koreanraichu/rcsb_pdb_2B10.fasta", "fasta")
first_record = next(record_iterator)
print(first_record.id)
print(first_record.description)
second_record = next(record_iterator)
print(second_record.id)
print(second_record.description)from Bio import SeqIO
first_record_2 = next(SeqIO.parse("/home/koreanraichu/rcsb_pdb_2B10.fasta", "fasta"))
print(first_record_2)위 코드와 아래 코드 둘 다 iteration을 활용하는 건 맞다.
2B10_1|Chains
2B10_1|Chains A, C|Cytochrome c peroxidase, mitochondrial|Saccharomyces cerevisiae (4932)
2B10_2|Chains
2B10_2|Chains B, D|Cytochrome c iso-1|Saccharomyces cerevisiae (4932)ID: 2B10_1|Chains
Name: 2B10_1|Chains
Description: 2B10_1|Chains A, C|Cytochrome c peroxidase, mitochondrial|Saccharomyces cerevisiae (4932)
Number of features: 0
Seq('TTPLVHVASVEKGRSYEDFQKVYNAIALKLREDDEYDNYIGYGPVLVRLAWHTS...QGL')위 코드는 next 수가 두 개라 앞에서 두 데이터+원하는 부분만 띄운 거고 아래는 판다스의 head()마냥 맨 위에걸 띄운거다. 아니 그러면 next를 일일이 쳐야되나요? for문 안됨? 있어봐요 해봅시다.
from Bio import SeqIO
record_iterator = SeqIO.parse("/home/koreanraichu/ls_orchid.fasta", "fasta")
for i in range(0,4):
record = next(record_iterator)
print(record.id)
print(record.description)gi|2765658|emb|Z78533.1|CIZ78533
gi|2765658|emb|Z78533.1|CIZ78533 C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA
gi|2765657|emb|Z78532.1|CCZ78532
gi|2765657|emb|Z78532.1|CCZ78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA
gi|2765656|emb|Z78531.1|CFZ78531
gi|2765656|emb|Z78531.1|CFZ78531 C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DNA
gi|2765655|emb|Z78530.1|CMZ78530
gi|2765655|emb|Z78530.1|CMZ78530 C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 DNA되는데요?
from Bio import SeqIO
record_iterator = SeqIO.parse("/home/koreanraichu/ls_orchid.fasta", "fasta")
i=0
while i<4
record = next(record_iterator)
print(record.id)
print(record.description)
i=i+1참고로 While문도 된다.
레코드 목록 뽑기
from Bio import SeqIO
records = list(SeqIO.parse("/home/koreanraichu/sequence.gb", "genbank"))
print(records)이게 무옵션 기본 코드이다.
print("First record:",records[0])
print("Last record:",records[-1])인덱싱도 된다.
first=records[0]
last=records[-1]
# 변수 안 주면 풀로 나온다...
print("First record ID:",first.id)
print("Last record ID:",last.id)인덱싱 하면서 특정 인자만 보는 것도 된다. (위 코드는 처음과 맨 마지막 데이터의 아이디를 보여주는 코드)
데이터 추출
from Bio import SeqIO
record_iterator = SeqIO.parse("/home/koreanraichu/ls_orchid.gbk", "genbank")
first_record = next(record_iterator)
# 첫 번째 레코드를 가져온다.
print(first_record.annotations) # Annotations
print(first_record.annotations.keys()) # Key
print(first_record.annotations.values()) # Values{'molecule_type': 'DNA', 'topology': 'linear', 'data_file_division': 'PLN', 'date': '30-NOV-2006', 'accessions': ['Z78533'], 'sequence_version': 1, 'gi': '2765658', 'keywords': ['5.8S ribosomal RNA', '5.8S rRNA gene', 'internal transcribed spacer', 'ITS1', 'ITS2'], 'source': 'Cypripedium irapeanum', 'organism': 'Cypripedium irapeanum', 'taxonomy': ['Eukaryota', 'Viridiplantae', 'Streptophyta', 'Embryophyta', 'Tracheophyta', 'Spermatophyta', 'Magnoliophyta', 'Liliopsida', 'Asparagales', 'Orchidaceae', 'Cypripedioideae', 'Cypripedium'], 'references': [Reference(title='Phylogenetics of the slipper orchids (Cypripedioideae: Orchidaceae): nuclear rDNA ITS sequences', ...), Reference(title='Direct Submission', ...)]}
# 그냥 주석을 출력했을 때
dict_keys(['molecule_type', 'topology', 'data_file_division', 'date', 'accessions', 'sequence_version', 'gi', 'keywords', 'source', 'organism', 'taxonomy', 'references'])
# 키 주세요
dict_values(['DNA', 'linear', 'PLN', '30-NOV-2006', ['Z78533'], 1, '2765658', ['5.8S ribosomal RNA', '5.8S rRNA gene', 'internal transcribed spacer', 'ITS1', 'ITS2'], 'Cypripedium irapeanum', 'Cypripedium irapeanum', ['Eukaryota', 'Viridiplantae', 'Streptophyta', 'Embryophyta', 'Tracheophyta', 'Spermatophyta', 'Magnoliophyta', 'Liliopsida', 'Asparagales', 'Orchidaceae', 'Cypripedioideae', 'Cypripedium'], [Reference(title='Phylogenetics of the slipper orchids (Cypripedioideae: Orchidaceae): nuclear rDNA ITS sequences', ...), Reference(title='Direct Submission', ...)]])
# 값 주세요Annotation은 저장 형태가 딕셔너리라 이렇게 나온다고... 딕셔너리 형태라는 건 Key:value로 저장되어 있기 때문에 키값이나 밸류값만 볼 수 있다는 얘기이기도 하다.
print(first_record.annotations["source"]) # Annotation+selector(키 중 하나를 선택)
Cypripedium irapeanum # 결과이런 식으로 키 중 하나를 선택해서 거기에 대한 밸류를 볼 수 있다.
# 아래 코드는 리스트업 코드이다.
all_species = []
for seq_record in record_iterator:
all_species.append(seq_record.annotations["organism"])
print(all_species)숫자만 맞으면 어레이 만들어도 된다.
all_species = [
seq_record.annotations["organism"]
for seq_record in SeqIO.parse("ls_orchid.gbk", "genbank")
]
print(all_species)for문에 던져버려도 되고.
수정
from Bio import SeqIO
record_iterator = SeqIO.parse("/home/koreanraichu/rcsb_pdb_2B10_mod.fasta", "fasta") # 불러와서(사본 만듦)
first_record = next(record_iterator)
first_record.id="SC_2B10_1" # ID를 바꿨다.
print(first_record.id)원본 파일에 손대기 좀 그래서 사본 만들어서 수정했다. 수정은 이런 식으로 하면 된다.
웹에서 받아오기
아 솔직치 다운받기 귀찮은데 아이디 알면 걍 웹에서 뜯어오면 안돼요? 됨.
파일 주세요 Genbank여...
from Bio import Entrez
from Bio import SeqIO
Entrez.email = "blackholekun@gmail.com"
with Entrez.efetch(
db="nucleotide", rettype="fasta", retmode="text", id="60391722"
) as handle:
seq_record = SeqIO.read(handle, "fasta")
print(seq_record)순순히 파일을 내놓는다면 유혈사태는 면할 것이다! (메일 내꺼임)
ID: AB018076.3
Name: AB018076.3
Description: AB018076.3 Homo sapiens hedgehog gene, complete cds
Number of features: 0
Seq('ATGTCTCCCGCCCGGCTCCGGCCCCGACTGCACTTCTGCCTGGTCCTGTTGCTG...ACC')그래서 받아옴
from Bio import Entrez
from Bio import SeqIO
Entrez.email = "blackholekun@gmail.com"
with Entrez.efetch(
db="nucleotide", rettype="gb", retmode="text", id="60391722"
) as handle:
seq_record = SeqIO.read(handle, "gb")
print(seq_record)파일 또 내놔!
ID: AB018076.3
Name: AB018076
Description: Homo sapiens hedgehog gene, complete cds
Number of features: 8
/molecule_type=DNA
/topology=linear
/data_file_division=PRI
/date=24-FEB-2005
/accessions=['AB018076', 'AB010092', 'AB018075']
/sequence_version=3
/keywords=['']
/source=Homo sapiens (human)
/organism=Homo sapiens
/taxonomy=['Eukaryota', 'Metazoa', 'Chordata', 'Craniata', 'Vertebrata', 'Euteleostomi', 'Mammalia', 'Eutheria', 'Euarchontoglires', 'Primates', 'Haplorrhini', 'Catarrhini', 'Hominidae', 'Homo']
/references=[Reference(title='Expression of Sonic hedgehog and its receptor Patched/Smoothened in human cancer cell lines and embryonic organs', ...), Reference(title='Direct Submission', ...)]
/comment=On or before Mar 1, 2005 this sequence version replaced AB010092.1,
AB018075.1, AB018076.2.
Seq('ATGTCTCCCGCCCGGCTCCGGCCCCGACTGCACTTCTGCCTGGTCCTGTTGCTG...ACC')아, 이건 genbank 포맷이다. 파스타랑 저거랑 다 된다. 단백질 이름이요? 아니 진짜 있는건데???
from Bio import Entrez
from Bio import SeqIO
Entrez.email = "blackholekun@gmail.com"
with Entrez.efetch(
db="nucleotide", rettype="gb", retmode="text", id="60391722,60391706,1519245148" # 여러개도 된다.
) as handle:
for seq_record in SeqIO.parse(handle, "gb"):
print(seq_record.description)Homo sapiens hedgehog gene, complete cds
Homo sapiens hedgehog gene, complete cds
Homo sapiens sonic hedgehog signaling molecule (SHH), transcript variant 1, mRNA아 물론 뭉텅이로 받는것도 된다.
파일 주세요 swissprot이여...
솔직히 이정도면 PDB랑 유니프롯이랑 TAIR랑 펍켐도 지원해줘야된다. 개발자 일해라.
from Bio import ExPASy
from Bio import SeqIO
with ExPASy.get_sprot_raw("O23729") as handle:
seq_record = SeqIO.read(handle, "swiss")
print(seq_record)스위스프롯에서는 이렇게 가져온다.
ID: O23729
Name: CHS3_BROFI
Description: RecName: Full=Chalcone synthase 3; EC=2.3.1.74; AltName: Full=Naringenin-chalcone synthase 3;
Database cross-references: EMBL:AF007097, SMR:O23729, PRIDE:O23729, UniPathway:UPA00154, GO:GO:0016210, GO:GO:0009813, Gene3D:3.40.47.10, InterPro:IPR012328, InterPro:IPR001099, InterPro:IPR018088, InterPro:IPR011141, InterPro:IPR016039, PANTHER:PTHR11877, Pfam:PF02797, Pfam:PF00195, PIRSF:PIRSF000451, SUPFAM:SSF53901, PROSITE:PS00441
Number of features: 2
/molecule_type=protein
/accessions=['O23729']
/protein_existence=2
/date=15-JUL-1999
/sequence_version=1
/date_last_sequence_update=01-JAN-1998
/date_last_annotation_update=10-FEB-2021
/entry_version=75
/gene_name=Name=CHS3;
/organism=Bromheadia finlaysoniana (Orchid)
/taxonomy=['Eukaryota', 'Viridiplantae', 'Streptophyta', 'Embryophyta', 'Tracheophyta', 'Spermatophyta', 'Magnoliopsida', 'Liliopsida', 'Asparagales', 'Orchidaceae', 'Epidendroideae', 'Vandeae', 'Adrorhizinae', 'Bromheadia']
/ncbi_taxid=['41205']
/comment=FUNCTION: The primary product of this enzyme is 4,2',4',6'- tetrahydroxychalcone (also termed naringenin-chalcone or chalcone) which can under specific conditions spontaneously isomerize into naringenin.
CATALYTIC ACTIVITY: Reaction=4-coumaroyl-CoA + 2 H(+) + 3 malonyl-CoA = 2',4,4',6'- tetrahydroxychalcone + 3 CO2 + 4 CoA; Xref=Rhea:RHEA:11128, ChEBI:CHEBI:15378, ChEBI:CHEBI:16526, ChEBI:CHEBI:57287, ChEBI:CHEBI:57355, ChEBI:CHEBI:57384, ChEBI:CHEBI:77645; EC=2.3.1.74; Evidence={ECO:0000255|PROSITE-ProRule:PRU10023};
PATHWAY: Secondary metabolite biosynthesis; flavonoid biosynthesis.
SIMILARITY: Belongs to the thiolase-like superfamily. Chalcone/stilbene synthases family. {ECO:0000305}.
/references=[Reference(title='Molecular cloning and sequence analysis of chalcone synthase cDNAs of Bromheadia finlaysoniana.', ...)]
/keywords=['Acyltransferase', 'Flavonoid biosynthesis', 'Transferase']
Seq('MAPAMEEIRQAQRAEGPAAVLAIGTSTPPNALYQADYPDYYFRITKSEHLTELK...GAE')뭔진 모르겠지만 일단 잘못했어요
참고로 저거 치니까 유니프롯(Uniprot) 나오길래 여기서 아이디 제공해주는건가 했는데 저기다가 유니프롯 아이디 넣으니까 됨.
from Bio import ExPASy
from Bio import SeqIO
with ExPASy.get_sprot_raw("Q14005") as handle:
seq_record = SeqIO.read(handle, "swiss")
print(seq_record.name)
print(seq_record.description)Q14005: Interleukin-16
IL16_HUMAN
RecName: Full=Pro-interleukin-16; Contains: RecName: Full=Interleukin-16; Short=IL-16; AltName: Full=Lymphocyte chemoattractant factor; Short=LCF;뭐야 왜 돼요 ㄷㄷ
딕셔너리화하기
뭐 하면 뭐 좋은갑지... 모르것다... (뇌클리어)
SeqIO.to_dict
from Bio import SeqIO
orchid_dict = SeqIO.to_dict(SeqIO.parse("/home/koreanraichu/sequence.gb", "genbank"))
print(orchid_dict.keys())
print(orchid_dict.values())dict_keys(['NM_000517.6'])
dict_values([SeqRecord(seq=Seq('ACTCTTCTGGTCCCCACAGACTCAGAGAGAACCCACCATGGTGCTGTCTCCTGC...GCA'), id='NM_000517.6', name='NM_000517', description='Homo sapiens hemoglobin subunit alpha 2 (HBA2), mRNA', dbxrefs=[])])이런 식으로 딕셔너리화할 수 있고
print(orchid_dict['2B10_1|Chains'].description)
2B10_1|Chains A, C|Cytochrome c peroxidase, mitochondrial|Saccharomyces cerevisiae (4932)이런 식으로 셀렉터 적용도 된다.
for record in SeqIO.parse("/home/koreanraichu/rcsb_pdb_2B10.fasta", "fasta"):
print(record.id, seguid(record.seq))SEGUID checksum 함수를 적용시켜 딕셔너리화 할 수도 있다는데...
2B10_1|Chains kSIi2w+CcP3y/k3bFRVhvvWtCtM
2B10_2|Chains SA4dH3bOSXcG43088Houu01TH2U이건 키가 일단 너무 저세상이다. InChl key 못지 않음...
seguid_dict = SeqIO.to_dict(SeqIO.parse("/home/koreanraichu/rcsb_pdb_2B10.fasta", "fasta"),lambda rec: seguid(rec.seq))
record = seguid_dict["kSIi2w+CcP3y/k3bFRVhvvWtCtM"]
print(record.id)
2B10_1|Chains그러니까 저 함수 적용하고 저 키를 다 외우고 있으면 찾는 게 가능하다는 얘기.
SeqIO.index
from Bio import SeqIO
orchid_dict = SeqIO.index("/home/koreanraichu/Octodon degus insulin.gb", "genbank")
print(orchid_dict.keys())
print(orchid_dict.values())KeysView(SeqIO.index('/home/koreanraichu/Octodon degus insulin.gb', 'genbank', alphabet=None, key_function=None))
ValuesView(SeqIO.index('/home/koreanraichu/Octodon degus insulin.gb', 'genbank', alphabet=None, key_function=None))예제 보면 리스트로 잘만 나오던데 나는 왜 저따구여?
쓰기
언제까지 긁어만 올텐가...
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
rec1 = SeqRecord(
Seq(
"MMYQQGCFAGGTVLRLAKDLAENNRGARVLVVCSEITAVTFRGPSETHLDSMVGQALFGD"
"GAGAVIVGSDPDLSVERPLYELVWTGATLLPDSEGAIDGHLREVGLTFHLLKDVPGLISK"
"NIEKSLKEAFTPLGISDWNSTFWIAHPGGPAILDQVEAKLGLKEEKMRATREVLSEYGNM"
"SSAC",
),
id="gi|14150838|gb|AAK54648.1|AF376133_1",
description="chalcone synthase [Cucumis sativus]",
)
rec2 = SeqRecord(
Seq(
"YPDYYFRITNREHKAELKEKFQRMCDKSMIKKRYMYLTEEILKENPSMCEYMAPSLDARQ"
"DMVVVEIPKLGKEAAVKAIKEWGQ",
),
id="gi|13919613|gb|AAK33142.1|",
description="chalcone synthase [Fragaria vesca subsp. bracteata]",
)
rec3 = SeqRecord(
Seq(
"MVTVEEFRRAQCAEGPATVMAIGTATPSNCVDQSTYPDYYFRITNSEHKVELKEKFKRMC"
"EKSMIKKRYMHLTEEILKENPNICAYMAPSLDARQDIVVVEVPKLGKEAAQKAIKEWGQP"
"KSKITHLVFCTTSGVDMPGCDYQLTKLLGLRPSVKRFMMYQQGCFAGGTVLRMAKDLAEN"
"NKGARVLVVCSEITAVTFRGPNDTHLDSLVGQALFGDGAAAVIIGSDPIPEVERPLFELV"
"SAAQTLLPDSEGAIDGHLREVGLTFHLLKDVPGLISKNIEKSLVEAFQPLGISDWNSLFW"
"IAHPGGPAILDQVELKLGLKQEKLKATRKVLSNYGNMSSACVLFILDEMRKASAKEGLGT"
"TGEGLEWGVLFGFGPGLTVETVVLHSVAT",
),
id="gi|13925890|gb|AAK49457.1|",
description="chalcone synthase [Nicotiana tabacum]",
)
my_records = [rec1, rec2, rec3]
from Bio import SeqIO
SeqIO.write(my_records, "/home/koreanraichu/example.fasta", "fasta")아니 이거 내가 IL로 트라이했는데 에러났음... 예제껀 잘됐는데 뭔 타입에러가 반기던데?
아무튼 이런 식으로 저장된다. (메시지 출력 따로 안했다)
from Bio import SeqIO
records = SeqIO.parse("/home/koreanraichu/sequence.gb", "genbank")
count = SeqIO.write(records, "/home/koreanraichu/my_example.fasta", "fasta")
print("Converted %i records" % count)
count2 = SeqIO.convert("/home/koreanraichu/sequence.gb", "genbank", "/home/koreanraichu/my_example2.fasta", "fasta")
print("Converted %i records" % count2)긁어오는 방법은 두 가지고 둘 다 된다.
from Bio import SeqIO
records = [rec.reverse_complement(id="rc_"+rec.id, description = "reverse complement")
for rec in SeqIO.parse("/home/koreanraichu/at5g40780.gb", "genbank")]
SeqIO.write(records, "/home/koreanraichu/at5g40780_rc.fasta", "fasta")DNA의 경우 reverse complement sequence로 저장하는 것도 된다. (단백질은 하지말자...)
'Coding > Python' 카테고리의 다른 글
| 번외편-코딩테스트 풀이 (0) | 2022.08.20 |
|---|---|
| Biopython으로 MSA 해보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 레코드 생성하고 만져보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 다뤄보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 가져오기 (0) | 2022.08.20 |
오늘껀 근데 하면서 나도 이해 못했음...
SeqRecord를 만들려면 뭘 또 모셔와야 하는데 바로
from Bio.SeqRecord import SeqRecord이놈이다.
이정도면 복잡한 거 돌리려면 아주 모셔오는데만 열댓줄 쓰게 생겼는데??? 텐서플로우나 저기 그 넘파이 판다스처럼 합쳐버리지 그러냐... (걔네는 일단 한 번 데려오면 다 써먹을 수 있음)
레코드 생성하기
이모지 넣고싶은데 이거 리눅스라 이모지 넣는 법을 모름... 아무튼 레코드는 이렇게 생성하면 된다.
from Bio.SeqRecord import SeqRecord
from Bio.Seq import Seq
EcoRI=Seq("GAATTC") # ECoRI sequence
ECoRI_r=SeqRecord(EcoRI) #로 레코드를 만들어보겠습니다
print(EcoRI)
print(ECoRI_r)그러면 어떻게 나오느냐
GAATTC # 시퀀스
ID: <unknown id> # 여기서부터 레코드
Name: <unknown name>
Description: <unknown description>
Number of features: 0
Seq('GAATTC')이렇게 나온다. 뭐가 많이 빠졌지만 일단 레코드 맞다. 여기다 넣고싶으면 일단 다음 문단으로 가시고... 아니 그럼 생성할 때 넣을 수 있나요? 예. 됩니다.
HaeIII_r=SeqRecord(HaeIII,id="RES_HAEIII",name="HaeIII restriction site",description="Restriction of HaeIII, cuts GG/CC")노션도 되는 wrap이 안되는 코드블록... 네이버 니네 점검하면 대체 뭐하냐... 아무튼 이렇게 만들 때 넣으면
ID: RES_HAEIII
Name: HaeIII restriction site
Description: Restriction of HaeIII, cuts GG/CC
Number of features: 0
Seq('GGCC')이렇게 생성된다.
레코드에 뭔가 넣기
아니 아까 만든거에 뭐 좀 어떻게 넣어주면 안됩니까? 아뇨 됩니다.
# 레코드에 정보를 추가해보자
ECoRI_r.id="RES_ECORI"
ECoRI_r.name="ECoRI restriction site"
ECoRI_r.description="Restriction site of ECoRI, cuts G/AATTC"
# ID랑 이름, description 정보를 추가했다.이렇게 필요한 정보를 넣어줄 수 있다. (참고: EcoRI이 맞는 표기다)
ID: RES_ECORI
Name: ECoRI restriction site
Description: Restriction site of ECoRI, cuts G/AATTC
Number of features: 0
Seq('GAATTC')제가 된다고 했잖아여...
HaeIII_r.annotations["notes"]="갑자기 해삼이 먹고싶다. " # annotation
HaeIII_r.letter_annotations['letter annotations']=['G','G/','C','C'] # letter annotations/notes=갑자기 해삼이 먹고싶다.
Per letter annotation for: letter annotations주석도 넣을 수 있다. 참고로 HaeIII은 Haemophilus influenzae biogroup aegyptius에서 유래한 제한효소. 본인은 해삼 혹은 해쓰리로 읽는다. (자매품 XbaI...)
FASTA에서 긁어오기
FASTA와 Genbank 파일에서 레코드를 긁어올 수도 있다. 후자쪽이 정보는 많음.
from Bio import SeqIO
record=SeqIO.read('/home/koreanraichu/rcsb_pdb_7PNN.fasta','fasta')
print(record)record=SeqIO.parse('/home/koreanraichu/rcsb_pdb_2B10.fasta','fasta')FASTA 파일에 하나 들어있으면 read, 두 개 이상이면 parse로 긁어야 한다. (두 개 이상인데 read로 긁으면 ValueError: More than one record found in handle가 당신을 반길 것이다)
print(record.id)
7PNN_1|Chainfor record2 in SeqIO.parse("/home/koreanraichu/rcsb_pdb_2B10.fasta","fasta"):
print(record2.id)
2B10_1|Chains
2B10_2|Chains긁는 난이도치고 접근은 되게 쉽다. 근데 parse로 긁어오면 for문 써야 나온다...
Genbank에서 긁어오기
record=SeqIO.read("/home/koreanraichu/sequence.gb",'genbank')
print(record)print(record.description)
Homo sapiens hemoglobin subunit alpha 2 (HBA2), mRNA얘는 리드로 파스고 없이 걍 긁어오면 접근이 바로 된다. 편한데?
Feature
시퀀스 레코드의 정보를 담고 있다고 하는데 뭔지 모르겠는 건 기분탓이 아니다. 여기서 볼 것은 .type와 .location, .qualifiers이다.
.type
for feature in record.features:
print(feature.type)
source
gene
CDS
sig_peptide
mat_peptide
mat_peptide
mat_peptide
regulatory
polyA_site얘도 for문 돌려야 나온다. 타입은 말 그대로 타입.
.location
for feature in record.features:
print(feature.location)
[0:432](+)
[0:432](+)
[41:371](+)
[41:113](+)
[113:200](+)
[206:293](+)
[299:368](+)
[413:419](+)
[431:432](+)Feature의 위치 정보. +는 뭐임?
.qualifiers
for feature in record.features:
print(feature.qualifiers)
OrderedDict([('organism', ['Octodon degus']), ('mol_type', ['mRNA']), ('db_xref', ['taxon:10160']), ('tissue_type', ['pancreas'])])
OrderedDict([('gene', ['insulin'])])
OrderedDict([('gene', ['insulin']), ('codon_start', ['1']), ('product', ['insulin']), ('protein_id', ['AAA40590.1']), ('translation', ['MAPWMHLLTVLALLALWGPNSVQAYSSQHLCGSNLVEALYMTCGRSGFYRPHDRRELEDLQVEQAELGLEAGGLQPSALEMILQKRGIVDQCCNNICTFNQLQNYCNVP'])])
OrderedDict([('gene', ['insulin'])])
OrderedDict([('gene', ['insulin']), ('product', ['insulin B-chain'])])
OrderedDict([('gene', ['insulin']), ('product', ['insulin C-peptide'])])
OrderedDict([('gene', ['insulin']), ('product', ['insulin A-chain'])])
OrderedDict([('regulatory_class', ['polyA_signal_sequence']), ('gene', ['insulin'])])
OrderedDict([('gene', ['insulin'])])Feature를 딕셔너리 형태로 저장해둔 것. Octodon degus는 데덴네의 모티브가 되기도 한 데구의 학명이다.
Position과 location
Feature의 위치 정보. Position은 0차원(점)이고 Location은 1차원(선)이다.

location은 포지션 두 개가 있어야된다. 여부터 여까지라는 얘기니께...
from Bio import SeqFeature
start_pos=SeqFeature.ExactPosition(15)
end_pos=SeqFeature.ExactPosition(30)
location=SeqFeature.FeatureLocation(start_pos, end_pos)
print(start_pos,end_pos,location)
15 30 [15:30]start_pos2 = SeqFeature.AfterPosition(1)
end_pos2 = SeqFeature.BetweenPosition(9, left=8, right=9)
my_location = SeqFeature.FeatureLocation(start_pos2, end_pos2)
print(start_pos2,end_pos2,my_location)
>1 (8^9) [>1:(8^9)]30쓰니까 에러뜸...
포지션이 명확할 때도 있고 명확하지 않을 때도 있다. 무슨 양자역학이냐... 슈뢰딩거의 포지션
start_pos2 = SeqFeature.AfterPosition(1)
end_pos2 = SeqFeature.BeforePosition(8)
my_location = SeqFeature.FeatureLocation(start_pos2, end_pos2)
print(start_pos2,end_pos2,my_location)
>1 <8 [>1:<8]참고로 애프터가 있다는 것은 비포가 있다는 얘기이기도 하다. (대충 펀쿨섹좌 짤)
실제 파일에 적용해보기
ID: NM_000517.6
Name: NM_000517
Description: Homo sapiens hemoglobin subunit alpha 2 (HBA2), mRNA
Number of features: 44
/molecule_type=mRNA
/topology=linear
/data_file_division=PRI
/date=06-SEP-2021
/accessions=['NM_000517']
/sequence_version=6
/keywords=['RefSeq', 'MANE Select']
/source=Homo sapiens (human)
/organism=Homo sapiens
/taxonomy=['Eukaryota', 'Metazoa', 'Chordata', 'Craniata', 'Vertebrata', 'Euteleostomi', 'Mammalia', 'Eutheria', 'Euarchontoglires', 'Primates', 'Haplorrhini', 'Catarrhini', 'Hominidae', 'Homo']
/references=[Reference(title='Molecular and Hematological Analysis of Alpha- and Beta-Thalassemia in a Cohort of Mexican Patients', ...), Reference(title='Molecular Characterization and Hematological Aspects of Hb E-Myanmar [beta26(B8)Glu-->Lys and beta65(E9)Lys-->Asn, HBB: c.[79G>A;198G>C]): A Novel beta-Thalassemic Hemoglobin Variant', ...), Reference(title='delta-Globin Chain Variants Associated with Decreased Hb A2 Levels: A National Reference Laboratory Experience', ...), Reference(title='Heterozygosity for the Novel HBA2: c.*91_*92delTA Polyadenylation Site Variant on the alpha2-Globin Gene Expanding the Genetic Spectrum of alpha-Thalassemia in Iran', ...), Reference(title='Identification and characterization of two novel and differentially expressed isoforms of human alpha2- and alpha1-globin genes', ...), Reference(title='Alpha-Thalassemia', ...), Reference(title="Cloning and complete nucleotide sequence of human 5'-alpha-globin gene", ...), Reference(title='A new abnormal human hemoglobin: Hb Prato (alpha 2 31 (B12) Arg leads to Ser beta 2)', ...), Reference(title='Hemoglobin Suan-Dok (alpha 2 109 (G16) Leu replaced by Arg beta 2): an unstable variant associated with alpha-thalassemia', ...), Reference(title='Hemoglobin Tarrant: alpha126(H9) Asp leads to Asn. A new hemoglobin variant in the alpha1beta1 contact region showing high oxygen affinity and reduced cooperativity', ...)]
/comment=REVIEWED REFSEQ: This record has been curated by NCBI staff. The
reference sequence was derived from V00493.1, Z84721.1 and
AI016696.1.
This sequence is a reference standard in the RefSeqGene project.
On Aug 3, 2018 this sequence version replaced NM_000517.5.
Summary: The human alpha globin gene cluster located on chromosome
16 spans about 30 kb and includes seven loci: 5'- zeta - pseudozeta
- mu - pseudoalpha-1 - alpha-2 - alpha-1 - theta - 3'. The alpha-2
(HBA2) and alpha-1 (HBA1) coding sequences are identical. These
genes differ slightly over the 5' untranslated regions and the
introns, but they differ significantly over the 3' untranslated
regions. Two alpha chains plus two beta chains constitute HbA,
which in normal adult life comprises about 97% of the total
hemoglobin; alpha chains combine with delta chains to constitute
HbA-2, which with HbF (fetal hemoglobin) makes up the remaining 3%
of adult hemoglobin. Alpha thalassemias result from deletions of
each of the alpha genes as well as deletions of both HBA2 and HBA1;
some nondeletion alpha thalassemias have also been reported.
[provided by RefSeq, Jul 2008].
Publication Note: This RefSeq record includes a subset of the
publications that are available for this gene. Please see the Gene
record to access additional publications.
COMPLETENESS: full length.
/structured_comment=OrderedDict([('Evidence-Data', OrderedDict([('Transcript exon combination', 'AI816040.1, AI815806.1 [ECO:0000332]'), ('RNAseq introns', 'single sample supports all introns SAMEA1965299, SAMEA1966682 [ECO:0000348]')])), ('RefSeq-Attributes', OrderedDict([('MANE Ensembl match', 'ENST00000251595.11/ ENSP00000251595.6'), ('RefSeq Select criteria', 'based on single protein-coding transcript')]))])
Seq('ACTCTTCTGGTCCCCACAGACTCAGAGAGAACCCACCATGGTGCTGTCTCCTGC...GCA')오늘의 시범조교. 뭐야 wrap 돌려줘요.
record=SeqIO.read("/home/koreanraichu/sequence.gb",'genbank') # 여기서 뭘 뽑아봅시다
index=575
for feature in record.features:
if index in feature:
print("%s, %s, %s" %(feature.type,feature.location,feature.qualifiers.get("db_xref")))여기서 575가 들어가는 feature를 찾아보자.
source, [0:576](+), ['taxon:9606']
gene, [0:576](+), ['GeneID:3040', 'HGNC:HGNC:4824', 'MIM:141850']
exon, [337:576](+), None아 그래서 이렇게 나온건가... (아직도 이해 못함)
SeqFeature로 시퀀스의 특정 부분만 잘라서 보기
from Bio.Seq import Seq
from Bio.SeqFeature import SeqFeature, FeatureLocation
at5g40780=Seq("MVAQAPHDDHQDDEKLAAARQKEIEDWLPITSSRNAKWWYSAFHNVTAMVGAGVLGLPYAMSQLGWGPGIAVLVLSWVITLYTLWQMVEMHEMVPGKRFDRYHELGQHAFGEKLGLYIVVPQQLIVEIGVCIVYMVTGGKSLKKFHELVCDDCKPIKLTYFIMIFASVHFVLSHLPNFNSISGVSLAAAVMSLSYSTIAWASSASKGVQEDVQYGYKAKTTAGTVFNFFSGLGDVAFAYAGHNVVLEIQATIPSTPEKPSKGPMWRGVIVAYIVVALCYFPVALVGYYIFGNGVEDNILMSLKKPAWLIATANIFVVIHVIGSYQIYAMPVFDMMETLLVKKLNFRPTTTLRFFVRNFYVAATMFVGMTFPFFGGLLAFFGGFAFAPTTYFLPCVIWLAIYKPKKYSLSWWANWVCIVFGLFLMVLSPIGGLRTIVIQAKGYKFYS")
# 단백질 시퀀스 왜 이렇게 길어 이거
feature = SeqFeature(FeatureLocation(0, 30), type="protein", strand=1)
feature_seq = at5g40780[feature.location.start:feature.location.end]
print(feature_seq)TAIR에서 돌아온 시범조교, LHT1의 단백질 시퀀스다. 뭐요. 파이참 wrap 일일이 켜기도 귀찮은데 걍 혀... 아무튼 feature라는 변수를 통해 30번째까지 볼 거라는 얘기다. strand가 -1이면 상보적인 시퀀스를 추출한다는데 저거 단백질이라 의미 없음.
feature_seq2 = feature.extract(at5g40780)
print(feature_seq2)Feature 변수 주고 extract해도 된다.
레코드 비교하기
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
record1=SeqRecord(Seq("GAATTC"), id="ECoR1",description="sticky")
record2=SeqRecord(Seq("GGCC"), id="HaeIII",description="blunt")
record3=SeqRecord(Seq("CTCGAG"), id="XhoI",description="sticky")
record4=SeqRecord(Seq("GATC"), id="DpnII",description="sticky")이번 시범조교는 얘네들이다.
레코드 비교? 그럼 다이렉트로 비교 되나요? 그러면 NotImplementedError: SeqRecord comparison is deliberately not implemented. Explicitly compare the attributes of interest. 에러가 당신을 반겨줄 것이다. 레코드 비교는 안에 든 걸 비교하는거지 레코드를 쌩으로 비교하라는 얘기가 아니다.
print(record1.description==record3.description)
TrueEcoRI과 XhoI의 description이 같으므로 참이다.
print(len(record1.seq)==len(record2.seq))
FalseEcoRI과 HaeIII의 시퀀스 길이는 다르므로 false.
포맷 만들기
아니 컴퓨터 포맷 아님.
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
record=SeqRecord(
Seq(
"ATGGTAGCTCAAGCTCCTCATGATGATCATCAGGATGATGAGAAATTAGCAGCAGCGAGACAAAAAGAGATCGAAGATTGGTTACCAATTACTTCATCAAGAAATGCAAAGTGGTGGTACTCTGCTTTTCACAATGTCACCGCCATGGTCGGTGCCGGAGTTCTTGGACTCCCTTACGCCATGTCTCAGCTCGGATGGGGACCGGGAATTGCAGTGTTGGTTTTGTCATGGGTCATAACACTATACACATTATGGCAAATGGTGGAAATGCATGAAATGGTTCCTGGAAAGCGTTTTGATCGTTACCATGAGCTCGGACAACACGCGTTTGGAGAAAAACTCGGTCTTTATATCGTTGTGCCGCAACAATTGATCGTTGAAATCGGTGTTTGCATCGTTTATATGGTCACTGGAGGCAAATCTTTAAAGAAATTTCATGAGCTTGTTTGTGATGATTGTAAACCAATCAAGCTTACTTATTTCATCATGATCTTTGCTTCGGTTCACTTCGTCCTCTCTCATCTTCCTAATTTCAATTCCATCTCCGGCGTTTCTCTTGCTGCTGCCGTTATGTCTCTCAGCTACTCAACAATCGCATGGGCATCATCAGCAAGCAAAGGTGTTCAAGAAGACGTTCAATACGGTTACAAAGCGAAAACAACAGCCGGTACGGTTTTCAATTTCTTCAGCGGTTTAGGTGATGTGGCATTTGCTTACGCGGGTCATAATGTGGTCCTTGAGATCCAAGCAACTATCCCTTCAACGCCTGAGAAACCCTCAAAAGGTCCCATGTGGAGAGGAGTCATCGTTGCTTACATCGTCGTAGCGCTCTGTTATTTCCCCGTGGCTCTCGTTGGATACTACATTTTCGGGAACGGAGTCGAAGATAATATTCTCATGTCACTTAAGAAACCGGCGTGGTTAATCGCCACGGCGAACATCTTCGTCGTGATCCATGTCATTGGTAGTTACCAGATATATGCAATGCCGGTATTTGATATGATGGAGACTTTATTGGTCAAGAAGCTAAATTTCAGACCAACCACAACTCTTCGGTTCTTTGTCCGTAATTTCTACGTTGCTGCAACTATGTTTGTTGGTATGACGTTTCCGTTCTTCGGTGGGCTTTTGGCGTTCTTTGGTGGATTCGCGTTTGCCCCAACCACATACTTCCTCCCTTGCGTCATTTGGTTAGCCATCTACAAACCCAAGAAATACAGCTTGTCTTGGTGGGCCAACTGGGTATGCATCGTGTTTGGTCTTTTCTTGATGGTCCTATCGCCAATTGGAGGGCTAAGGACAATCGTTATTCAAGCAAAAGGATACAAGTTTTACTCATAA"),
id="at5g40780|LHT1",
description="Lysine-histidine transporter 1|Arabidopsis thaliana")
print(record.format("fasta"))이런 식으로 입력하면
>at5g40780|LHT1 Lysine-histidine transporter 1|Arabidopsis thaliana
ATGGTAGCTCAAGCTCCTCATGATGATCATCAGGATGATGAGAAATTAGCAGCAGCGAGA
CAAAAAGAGATCGAAGATTGGTTACCAATTACTTCATCAAGAAATGCAAAGTGGTGGTAC
TCTGCTTTTCACAATGTCACCGCCATGGTCGGTGCCGGAGTTCTTGGACTCCCTTACGCC
ATGTCTCAGCTCGGATGGGGACCGGGAATTGCAGTGTTGGTTTTGTCATGGGTCATAACA
CTATACACATTATGGCAAATGGTGGAAATGCATGAAATGGTTCCTGGAAAGCGTTTTGAT
CGTTACCATGAGCTCGGACAACACGCGTTTGGAGAAAAACTCGGTCTTTATATCGTTGTG
CCGCAACAATTGATCGTTGAAATCGGTGTTTGCATCGTTTATATGGTCACTGGAGGCAAA
TCTTTAAAGAAATTTCATGAGCTTGTTTGTGATGATTGTAAACCAATCAAGCTTACTTAT
TTCATCATGATCTTTGCTTCGGTTCACTTCGTCCTCTCTCATCTTCCTAATTTCAATTCC
ATCTCCGGCGTTTCTCTTGCTGCTGCCGTTATGTCTCTCAGCTACTCAACAATCGCATGG
GCATCATCAGCAAGCAAAGGTGTTCAAGAAGACGTTCAATACGGTTACAAAGCGAAAACA
ACAGCCGGTACGGTTTTCAATTTCTTCAGCGGTTTAGGTGATGTGGCATTTGCTTACGCG
GGTCATAATGTGGTCCTTGAGATCCAAGCAACTATCCCTTCAACGCCTGAGAAACCCTCA
AAAGGTCCCATGTGGAGAGGAGTCATCGTTGCTTACATCGTCGTAGCGCTCTGTTATTTC
CCCGTGGCTCTCGTTGGATACTACATTTTCGGGAACGGAGTCGAAGATAATATTCTCATG
TCACTTAAGAAACCGGCGTGGTTAATCGCCACGGCGAACATCTTCGTCGTGATCCATGTC
ATTGGTAGTTACCAGATATATGCAATGCCGGTATTTGATATGATGGAGACTTTATTGGTC
AAGAAGCTAAATTTCAGACCAACCACAACTCTTCGGTTCTTTGTCCGTAATTTCTACGTT
GCTGCAACTATGTTTGTTGGTATGACGTTTCCGTTCTTCGGTGGGCTTTTGGCGTTCTTT
GGTGGATTCGCGTTTGCCCCAACCACATACTTCCTCCCTTGCGTCATTTGGTTAGCCATC
TACAAACCCAAGAAATACAGCTTGTCTTGGTGGGCCAACTGGGTATGCATCGTGTTTGGT
CTTTTCTTGATGGTCCTATCGCCAATTGGAGGGCTAAGGACAATCGTTATTCAAGCAAAA
GGATACAAGTTTTACTCATAA이렇게 FASTA 포맷이 된다.
슬라이싱
feature도 slicing이 된다. 어째서인지는 모르겠으나...
from Bio import SeqIO
record = SeqIO.read("/home/koreanraichu/sequence.gb", "genbank")
print(record.features[20])이건 근데 인덱싱 아니냐.
sub_record = record[0:5]
print(sub_record)
ID: NM_000517.6
Name: NM_000517
Description: Homo sapiens hemoglobin subunit alpha 2 (HBA2), mRNA
Number of features: 0
/molecule_type=mRNA
Seq('ACTCT')아무튼 레코드에서 0부터 5까지 뽑아보았다. 염기 6개가 나온다.
record = SeqIO.read("/home/koreanraichu/sequence.gb", "genbank")
sub_record = record[0:576]
print(sub_record.features[0])범위를 풀로 바꾸고 0번째 feature를 봤다.
type: source
location: [0:576](+)
qualifiers:
Key: chromosome, Value: ['16']
Key: db_xref, Value: ['taxon:9606']
Key: map, Value: ['16p13.3']
Key: mol_type, Value: ['mRNA']
Key: organism, Value: ['Homo sapiens']그러면 이렇게 나옵니다.
print(sub_record.format("genbank"))
print(sub_record.format("fasta"))위 코드를 이용해 출력 포맷을 바꿀 수도 있다.
>NM_000517.6 Homo sapiens hemoglobin subunit alpha 2 (HBA2), mRNA
ACTCTTCTGGTCCCCACAGA
# FASTALOCUS NM_000517 20 bp mRNA UNK 01-JAN-1980
DEFINITION Homo sapiens hemoglobin subunit alpha 2 (HBA2), mRNA.
ACCESSION NM_000517
VERSION NM_000517.6
KEYWORDS .
SOURCE .
ORGANISM .
.
FEATURES Location/Qualifiers
ORIGIN
1 actcttctgg tccccacaga
//
# Genbank위는 FASTA, 아래는 Genbank 포맷.
'Coding > Python' 카테고리의 다른 글
| 번외편-코딩테스트 풀이 (0) | 2022.08.20 |
|---|---|
| Biopython으로 MSA 해보기 (0) | 2022.08.20 |
| Biopython SeqIO 써보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 다뤄보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 가져오기 (0) | 2022.08.20 |
전에요? 아니 그건 가져온거고.
시퀀스도 텍스트나 리스트처럼 인덱싱과 슬라이싱이 된다. 방법도 똑같다. 그래서 그건 생략할 예정임... 그리고 그거 아니어도 분량이 많아요 또.
.count()
특정 문자열이 몇 개인지 세 준다. 뭐 시퀀스에서 아데닌이나 구아닌 갯수 세주고 그런거다. 이걸 이용하면 특정 단백질 시퀀스에서 특정 아미노산(카테고리면 겁나 세야된다...)이 차지하는 비율도 볼 수 있다.
쉬어가는 코너-Primer에서 GC함량 구하기
아 이거 중요합니다. Primer 만들 때 중요한 요소 중 하나가 GC 함량이다. 참고로 여기서는 두 가지 방법으로 구해볼건데 첫번째는 .count()를 이용해 G와 C의 수를 세서 전체 DNA 염기 수로 나누는 거고, 두번째는 바이오파이썬의 모듈을 이용하는 방법이다.
GC_cont=(primer.count('C')+primer.count('G'))/len(primer)
print(GC_cont*100,"%")from Bio.SeqUtils import GC
primer=Seq('CAGCAAGCAAAGGTGTTCAA')
print(GC(primer),"%")위는 직접 계산하는 방법이고 아래는 모듈을 사용했다. 모듈은 쓰려면 또 모셔와야 한다.
시퀀스에 시퀀스를 더하면 1+1인가요
시퀀스 그냥 +로 갖다 붙이거나 리스트업된 거 for문으로 붙이거나 join()쓰거나... 일단 for와 join 보고 갑시다.
seq_list=[Seq('ATCC'),Seq('ATAT'),Seq('TNNN')]
add = Seq("")
for i in seq_list:
add += i
print(add)seq_list=[Seq('ATCC'),Seq('ATAT'),Seq('TNNN')]
spacer=Seq('N'*5)
print(spacer.join(seq_list))
돌연변이 만들기
시퀀스 데이터는 튜플마냥 안에 있는 내용물을 수정할 수 없다. 수정하려면 또 다른 모듈을 모셔와야 한다.
from Bio.Seq import Seq
from Bio.Seq import MutableSeq #이걸 불러와서
my_seq=Seq("GAATTC")
mutable_seq=MutableSeq(my_seq) #적용해줘야 한다
mutable_seq[0]="A"
print(mutable_seq)Mutableseq에 던져둔 것은 타입도
<class 'Bio.Seq.MutableSeq'>로 바뀐다. 일반 시퀀스는 Seq.
전사번역
입력한 시퀀스를 mRNA로 전사도 해 주고, 번역도 된다. 물론 다이렉트로 된다.
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqUtils import GC
at5g40780=Seq('ATGGTAGCTCAAGCTCCTCATGATGATCATCAGGATGATGAGAAATTAGCAGCAGCGAGACAAAAAGAGATCGAAGATTGGTTACCAATTACTTCATCAAGAAATGCAAAGTGGTGGTACTCTGCTTTTCACAATGTCACCGCCATGGTCGGTGCCGGAGTTCTTGGACTCCCTTACGCCATGTCTCAGCTCGGATGGGGACCGGGAATTGCAGTGTTGGTTTTGTCATGGGTCATAACACTATACACATTATGGCAAATGGTGGAAATGCATGAAATGGTTCCTGGAAAGCGTTTTGATCGTTACCATGAGCTCGGACAACACGCGTTTGGAGAAAAACTCGGTCTTTATATCGTTGTGCCGCAACAATTGATCGTTGAAATCGGTGTTTGCATCGTTTATATGGTCACTGGAGGCAAATCTTTAAAGAAATTTCATGAGCTTGTTTGTGATGATTGTAAACCAATCAAGCTTACTTATTTCATCATGATCTTTGCTTCGGTTCACTTCGTCCTCTCTCATCTTCCTAATTTCAATTCCATCTCCGGCGTTTCTCTTGCTGCTGCCGTTATGTCTCTCAGCTACTCAACAATCGCATGGGCATCATCAGCAAGCAAAGGTGTTCAAGAAGACGTTCAATACGGTTACAAAGCGAAAACAACAGCCGGTACGGTTTTCAATTTCTTCAGCGGTTTAGGTGATGTGGCATTTGCTTACGCGGGTCATAATGTGGTCCTTGAGATCCAAGCAACTATCCCTTCAACGCCTGAGAAACCCTCAAAAGGTCCCATGTGGAGAGGAGTCATCGTTGCTTACATCGTCGTAGCGCTCTGTTATTTCCCCGTGGCTCTCGTTGGATACTACATTTTCGGGAACGGAGTCGAAGATAATATTCTCATGTCACTTAAGAAACCGGCGTGGTTAATCGCCACGGCGAACATCTTCGTCGTGATCCATGTCATTGGTAGTTACCAGATATATGCAATGCCGGTATTTGATATGATGGAGACTTTATTGGTCAAGAAGCTAAATTTCAGACCAACCACAACTCTTCGGTTCTTTGTCCGTAATTTCTACGTTGCTGCAACTATGTTTGTTGGTATGACGTTTCCGTTCTTCGGTGGGCTTTTGGCGTTCTTTGGTGGATTCGCGTTTGCCCCAACCACATACTTCCTCCCTTGCGTCATTTGGTTAGCCATCTACAAACCCAAGAAATACAGCTTGTCTTGGTGGGCCAACTGGGTATGCATCGTGTTTGGTCTTTTCTTGATGGTCCTATCGCCAATTGGAGGGCTAAGGACAATCGTTATTCAAGCAAAAGGATACAAGTTTTACTCATAA')
at5g40780_rc=at5g40780.reverse_complement()
# DNA와 상보적인 sequence를 만들었다.
at5g40780_mrna=at5g40780.transcribe()
at5g40780_mrna_rc=at5g40780_mrna.reverse_complement()
# 여기 RNA 추가요!
print(at5g40780_mrna)
AUGGUAGCUCAAGCUCCUCAUGAUGAUCAUCAGGAUGAUGAGAAAUUAGCAGCAGCGAGACAAAAAGAGAUCGAAGAUUGGUUACCAAUUACUUCAUCAAGAAAUGCAAAGUGGUGGUACUCUGCUUUUCACAAUGUCACCGCCAUGGUCGGUGCCGGAGUUCUUGGACUCCCUUACGCCAUGUCUCAGCUCGGAUGGGGACCGGGAAUUGCAGUGUUGGUUUUGUCAUGGGUCAUAACACUAUACACAUUAUGGCAAAUGGUGGAAAUGCAUGAAAUGGUUCCUGGAAAGCGUUUUGAUCGUUACCAUGAGCUCGGACAACACGCGUUUGGAGAAAAACUCGGUCUUUAUAUCGUUGUGCCGCAACAAUUGAUCGUUGAAAUCGGUGUUUGCAUCGUUUAUAUGGUCACUGGAGGCAAAUCUUUAAAGAAAUUUCAUGAGCUUGUUUGUGAUGAUUGUAAACCAAUCAAGCUUACUUAUUUCAUCAUGAUCUUUGCUUCGGUUCACUUCGUCCUCUCUCAUCUUCCUAAUUUCAAUUCCAUCUCCGGCGUUUCUCUUGCUGCUGCCGUUAUGUCUCUCAGCUACUCAACAAUCGCAUGGGCAUCAUCAGCAAGCAAAGGUGUUCAAGAAGACGUUCAAUACGGUUACAAAGCGAAAACAACAGCCGGUACGGUUUUCAAUUUCUUCAGCGGUUUAGGUGAUGUGGCAUUUGCUUACGCGGGUCAUAAUGUGGUCCUUGAGAUCCAAGCAACUAUCCCUUCAACGCCUGAGAAACCCUCAAAAGGUCCCAUGUGGAGAGGAGUCAUCGUUGCUUACAUCGUCGUAGCGCUCUGUUAUUUCCCCGUGGCUCUCGUUGGAUACUACAUUUUCGGGAACGGAGUCGAAGAUAAUAUUCUCAUGUCACUUAAGAAACCGGCGUGGUUAAUCGCCACGGCGAACAUCUUCGUCGUGAUCCAUGUCAUUGGUAGUUACCAGAUAUAUGCAAUGCCGGUAUUUGAUAUGAUGGAGACUUUAUUGGUCAAGAAGCUAAAUUUCAGACCAACCACAACUCUUCGGUUCUUUGUCCGUAAUUUCUACGUUGCUGCAACUAUGUUUGUUGGUAUGACGUUUCCGUUCUUCGGUGGGCUUUUGGCGUUCUUUGGUGGAUUCGCGUUUGCCCCAACCACAUACUUCCUCCCUUGCGUCAUUUGGUUAGCCAUCUACAAACCCAAGAAAUACAGCUUGUCUUGGUGGGCCAACUGGGUAUGCAUCGUGUUUGGUCUUUUCUUGAUGGUCCUAUCGCCAAUUGGAGGGCUAAGGACAAUCGUUAUUCAAGCAAAAGGAUACAAGUUUUACUCAUAA.transcribe()를 이용하면 전사도 해 준다. 예시에 있는 시퀀스는 at5g40780(의 CDS).
from Bio.Seq import Seq
at5g40780=Seq('ATGGTAGCTCAAGCTCCTCATGATGATCATCAGGATGATGAGAAATTAGCAGCAGCGAGACAAAAAGAGATCGAAGATTGGTTACCAATTACTTCATCAAGAAATGCAAAGTGGTGGTACTCTGCTTTTCACAATGTCACCGCCATGGTCGGTGCCGGAGTTCTTGGACTCCCTTACGCCATGTCTCAGCTCGGATGGGGACCGGGAATTGCAGTGTTGGTTTTGTCATGGGTCATAACACTATACACATTATGGCAAATGGTGGAAATGCATGAAATGGTTCCTGGAAAGCGTTTTGATCGTTACCATGAGCTCGGACAACACGCGTTTGGAGAAAAACTCGGTCTTTATATCGTTGTGCCGCAACAATTGATCGTTGAAATCGGTGTTTGCATCGTTTATATGGTCACTGGAGGCAAATCTTTAAAGAAATTTCATGAGCTTGTTTGTGATGATTGTAAACCAATCAAGCTTACTTATTTCATCATGATCTTTGCTTCGGTTCACTTCGTCCTCTCTCATCTTCCTAATTTCAATTCCATCTCCGGCGTTTCTCTTGCTGCTGCCGTTATGTCTCTCAGCTACTCAACAATCGCATGGGCATCATCAGCAAGCAAAGGTGTTCAAGAAGACGTTCAATACGGTTACAAAGCGAAAACAACAGCCGGTACGGTTTTCAATTTCTTCAGCGGTTTAGGTGATGTGGCATTTGCTTACGCGGGTCATAATGTGGTCCTTGAGATCCAAGCAACTATCCCTTCAACGCCTGAGAAACCCTCAAAAGGTCCCATGTGGAGAGGAGTCATCGTTGCTTACATCGTCGTAGCGCTCTGTTATTTCCCCGTGGCTCTCGTTGGATACTACATTTTCGGGAACGGAGTCGAAGATAATATTCTCATGTCACTTAAGAAACCGGCGTGGTTAATCGCCACGGCGAACATCTTCGTCGTGATCCATGTCATTGGTAGTTACCAGATATATGCAATGCCGGTATTTGATATGATGGAGACTTTATTGGTCAAGAAGCTAAATTTCAGACCAACCACAACTCTTCGGTTCTTTGTCCGTAATTTCTACGTTGCTGCAACTATGTTTGTTGGTATGACGTTTCCGTTCTTCGGTGGGCTTTTGGCGTTCTTTGGTGGATTCGCGTTTGCCCCAACCACATACTTCCTCCCTTGCGTCATTTGGTTAGCCATCTACAAACCCAAGAAATACAGCTTGTCTTGGTGGGCCAACTGGGTATGCATCGTGTTTGGTCTTTTCTTGATGGTCCTATCGCCAATTGGAGGGCTAAGGACAATCGTTATTCAAGCAAAAGGATACAAGTTTTACTCATAA')
# DNA sequence
at5g40780_mrna=at5g40780.transcribe()
# Transcription
print(at5g40780_mrna)
AUGGUAGCUCAAGCUCCUCAUGAUGAUCAUCAGGAUGAUGAGAAAUUAGCAGCAGCGAGACAAAAAGAGAUCGAAGAUUGGUUACCAAUUACUUCAUCAAGAAAUGCAAAGUGGUGGUACUCUGCUUUUCACAAUGUCACCGCCAUGGUCGGUGCCGGAGUUCUUGGACUCCCUUACGCCAUGUCUCAGCUCGGAUGGGGACCGGGAAUUGCAGUGUUGGUUUUGUCAUGGGUCAUAACACUAUACACAUUAUGGCAAAUGGUGGAAAUGCAUGAAAUGGUUCCUGGAAAGCGUUUUGAUCGUUACCAUGAGCUCGGACAACACGCGUUUGGAGAAAAACUCGGUCUUUAUAUCGUUGUGCCGCAACAAUUGAUCGUUGAAAUCGGUGUUUGCAUCGUUUAUAUGGUCACUGGAGGCAAAUCUUUAAAGAAAUUUCAUGAGCUUGUUUGUGAUGAUUGUAAACCAAUCAAGCUUACUUAUUUCAUCAUGAUCUUUGCUUCGGUUCACUUCGUCCUCUCUCAUCUUCCUAAUUUCAAUUCCAUCUCCGGCGUUUCUCUUGCUGCUGCCGUUAUGUCUCUCAGCUACUCAACAAUCGCAUGGGCAUCAUCAGCAAGCAAAGGUGUUCAAGAAGACGUUCAAUACGGUUACAAAGCGAAAACAACAGCCGGUACGGUUUUCAAUUUCUUCAGCGGUUUAGGUGAUGUGGCAUUUGCUUACGCGGGUCAUAAUGUGGUCCUUGAGAUCCAAGCAACUAUCCCUUCAACGCCUGAGAAACCCUCAAAAGGUCCCAUGUGGAGAGGAGUCAUCGUUGCUUACAUCGUCGUAGCGCUCUGUUAUUUCCCCGUGGCUCUCGUUGGAUACUACAUUUUCGGGAACGGAGUCGAAGAUAAUAUUCUCAUGUCACUUAAGAAACCGGCGUGGUUAAUCGCCACGGCGAACAUCUUCGUCGUGAUCCAUGUCAUUGGUAGUUACCAGAUAUAUGCAAUGCCGGUAUUUGAUAUGAUGGAGACUUUAUUGGUCAAGAAGCUAAAUUUCAGACCAACCACAACUCUUCGGUUCUUUGUCCGUAAUUUCUACGUUGCUGCAACUAUGUUUGUUGGUAUGACGUUUCCGUUCUUCGGUGGGCUUUUGGCGUUCUUUGGUGGAUUCGCGUUUGCCCCAACCACAUACUUCCUCCCUUGCGUCAUUUGGUUAGCCAUCUACAAACCCAAGAAAUACAGCUUGUCUUGGUGGGCCAACUGGGUAUGCAUCGUGUUUGGUCUUUUCUUGAUGGUCCUAUCGCCAAUUGGAGGGCUAAGGACAAUCGUUAUUCAAGCAAAAGGAUACAAGUUUUACUCAUAA.translate()는 번역이다. (*은 종결코돈)
전사는 그냥 뭐 전사했구나 하고 넘어가도 되는데, 번역은 옵션이 좀 있으니 일단 보고 가자.
to_stop=True: 처음 만나는 종결코돈에서 멈춘다
from Bio.Seq import Seq
at5g40780_mrna=Seq('AUGGUAGCUCAAGCUCCUCAUGAUGAUCAUCAGGAUGAUGAGAAAUUAGCAGCAGCGAGACAAAAAGAGAUCGAAGAUUGGUUACCAAUUACUUCAUCAAGAAAUGCAAAGUGGUGGUACUCUGCUUUUCACAAUGUCACCGCCAUGGUCGGUGCCGGAGUUCUUGGACUCCCUUACGCCAUGUCUCAGCUCGGAUGGGGACCGGGAAUUGCAGUGUUGGUUUUGUCAUGGGUCAUAACACUAUACACAUUAUGGCAAAUGGUGGAAAUGCAUGAAAUGGUUCCUGGAAAGCGUUUUGAUCGUUACCAUGAGCUCGGACAACACGCGUUUGGAGAAAAACUCGGUCUUUAUAUCGUUGUGCCGCAACAAUUGAUCGUUGAAAUCGGUGUUUGCAUCGUUUAUAUGGUCACUGGAGGCAAAUCUUUAAAGAAAUUUCAUGAGCUUGUUUGUGAUGAUUGUAAACCAAUCAAGCUUACUUAUUUCAUCAUGAUCUUUGCUUCGGUUCACUUCGUCCUCUCUCAUCUUCCUAAUUUCAAUUCCAUCUCCGGCGUUUCUCUUGCUGCUGCCGUUAUGUCUCUCAGCUACUCAACAAUCGCAUGGGCAUCAUCAGCAAGCAAAGGUGUUCAAGAAGACGUUCAAUACGGUUACAAAGCGAAAACAACAGCCGGUACGGUUUUCAAUUUCUUCAGCGGUUUAGGUGAUGUGGCAUUUGCUUACGCGGGUCAUAAUGUGGUCCUUGAGAUCCAAGCAACUAUCCCUUCAACGCCUGAGAAACCCUCAAAAGGUCCCAUGUGGAGAGGAGUCAUCGUUGCUUACAUCGUCGUAGCGCUCUGUUAUUUCCCCGUGGCUCUCGUUGGAUACUACAUUUUCGGGAACGGAGUCGAAGAUAAUAUUCUCAUGUCACUUAAGAAACCGGCGUGGUUAAUCGCCACGGCGAACAUCUUCGUCGUGAUCCAUGUCAUUGGUAGUUACCAGAUAUAUGCAAUGCCGGUAUUUGAUAUGAUGGAGACUUUAUUGGUCAAGAAGCUAAAUUUCAGACCAACCACAACUCUUCGGUUCUUUGUCCGUAAUUUCUACGUUGCUGCAACUAUGUUUGUUGGUAUGACGUUUCCGUUCUUCGGUGGGCUUUUGGCGUUCUUUGGUGGAUUCGCGUUUGCCCCAACCACAUACUUCCUCCCUUGCGUCAUUUGGUUAGCCAUCUACAAACCCAAGAAAUACAGCUUGUCUUGGUGGGCCAACUGGGUAUGCAUCGUGUUUGGUCUUUUCUUGAUGGUCCUAUCGCCAAUUGGAGGGCUAAGGACAAUCGUUAUUCAAGCAAAAGGAUACAAGUUUUACUCAUAA')
# 이건 mRNA 시퀀스고
at5g40780_protein=at5g40780_mrna.translate(to_stop=True)
#번역했다.
print(at5g40780_protein)
MVAQAPHDDHQDDEKLAAARQKEIEDWLPITSSRNAKWWYSAFHNVTAMVGAGVLGLPYAMSQLGWGPGIAVLVLSWVITLYTLWQMVEMHEMVPGKRFDRYHELGQHAFGEKLGLYIVVPQQLIVEIGVCIVYMVTGGKSLKKFHELVCDDCKPIKLTYFIMIFASVHFVLSHLPNFNSISGVSLAAAVMSLSYSTIAWASSASKGVQEDVQYGYKAKTTAGTVFNFFSGLGDVAFAYAGHNVVLEIQATIPSTPEKPSKGPMWRGVIVAYIVVALCYFPVALVGYYIFGNGVEDNILMSLKKPAWLIATANIFVVIHVIGSYQIYAMPVFDMMETLLVKKLNFRPTTTLRFFVRNFYVAATMFVGMTFPFFGGLLAFFGGFAFAPTTYFLPCVIWLAIYKPKKYSLSWWANWVCIVFGLFLMVLSPIGGLRTIVIQAKGYKFYS
table=2: 아래 사이트에서 제공하는 코돈 테이블로 번역할 수 있다. (기본은 1번 스탠다드)
https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
The Genetic Codes Compiled by Andrzej (Anjay) Elzanowski and Jim Ostell at National Center for Biotechnology Information (NCBI), Bethesda, Maryland, U.S.A. Last update of the Genetic Codes: Jan. 7, 2019 NCBI takes great care to ensure that the translation
www.ncbi.nlm.nih.gov
at5g40780_protein=at5g40780_mrna.translate(table=2)
MVAQAPHDDHQDDEKLAAA*QKEIEDWLPITSS*NAKWWYSAFHNVTAMVGAGVLGLPYAMSQLGWGPGIAVLVLSWVMTLYTLWQMVEMHEMVPGKRFDRYHELGQHAFGEKLGLYIVVPQQLIVEIGVCIVYMVTGGKSLKKFHELVCDDCKPIKLTYFIMIFASVHFVLSHLPNFNSISGVSLAAAVMSLSYSTIAWASSASKGVQEDVQYGYKAKTTAGTVFNFFSGLGDVAFAYAGHNVVLEIQATIPSTPEKPSKGPMW*GVIVAYIVVALCYFPVALVGYYIFGNGVEDNILMSLKKPAWLIATANIFVVIHVIGSYQMYAMPVFDMMETLLVKKLNF*PTTTLRFFVRNFYVAATMFVGMTFPFFGGLLAFFGGFAFAPTTYFLPCVIWLAIYKPKKYSLSWWANWVCIVFGLFLMVLSPIGGL*TIVIQAKGYKFYS*위 CDS를 table 2(Vertebrate mitochondria)로 설정하고 번역한 결과. *은 종결 코돈이다.
stop_symbol="@": 종결코돈의 기호를 @로 바꾼다.
at5g40780_protein=at5g40780_mrna.translate(table=2,stop_symbol="-")
MVAQAPHDDHQDDEKLAAA-QKEIEDWLPITSS-NAKWWYSAFHNVTAMVGAGVLGLPYAMSQLGWGPGIAVLVLSWVMTLYTLWQMVEMHEMVPGKRFDRYHELGQHAFGEKLGLYIVVPQQLIVEIGVCIVYMVTGGKSLKKFHELVCDDCKPIKLTYFIMIFASVHFVLSHLPNFNSISGVSLAAAVMSLSYSTIAWASSASKGVQEDVQYGYKAKTTAGTVFNFFSGLGDVAFAYAGHNVVLEIQATIPSTPEKPSKGPMW-GVIVAYIVVALCYFPVALVGYYIFGNGVEDNILMSLKKPAWLIATANIFVVIHVIGSYQMYAMPVFDMMETLLVKKLNF-PTTTLRFFVRNFYVAATMFVGMTFPFFGGLLAFFGGFAFAPTTYFLPCVIWLAIYKPKKYSLSWWANWVCIVFGLFLMVLSPIGGL-TIVIQAKGYKFYS-위 예시에서는 -로 바꿨다.
글자 다이렉트로 전사번역하기
시퀀스도 글자긴 한데 str과 다른 점이 있다면 상보적인 시퀀스를 만들 수 있다는 것이다. 근데 그렇다고 헐 시퀀스 안만들었다! 망했다! 이건 아니고...
from Bio.Seq import reverse_complement, transcribe, back_transcribe, translate #질문있는데 이걸 꼭 이렇게 불러야겠니
insulin="GCATTCTGAGGCATTCTCTAACAGGTTCTCGACCCTCCGCCATGGCCCCGTGGATGCATCTCCTCACCGTGCTGGCCCTGCTGGCCCTCTGGGGACCCAACTCTGTTCAGGCCTATTCCAGCCAGCACCTGTGCGGCTCCAACCTAGTGGAGGCACTGTACATGACATGTGGACGGAGTGGCTTCTATAGACCCCACGACCGCCGAGAGCTGGAGGACCTCCAGGTGGAGCAGGCAGAACTGGGTCTGGAGGCAGGCGGCCTGCAGCCTTCGGCCCTGGAGATGATTCTGCAGAAGCGCGGCATTGTGGATCAGTGCTGTAATAACATTTGCACATTTAACCAGCTGCAGAACTACTGCAATGTCCCTTAGACACCTGCCTTGGGCCTGGCCTGCTGCTCTGCCCTGGCAACCAATAAACCCCTTGAATGAG"
#Wrap 알아서 켜주면 안되냐고...
insulin_rc=reverse_complement(insulin)
insulin_mrna=transcribe(insulin)
insulin_prot=translate(insulin)
# 순서대로 reverse complement/전사/번역
print(insulin_rc)
print(insulin_mrna)
print(insulin_prot)시퀀스때랑 불러오는 건 다르지만 어쨌든 된다.
코돈 테이블 보기
내 블로그 아미노산 배경화면에도 있긴 한데 그걸 누가 외웁니까... 아무튼 그렇다.
from Bio.Data import CodonTable
standard_table = CodonTable.unambiguous_dna_by_name["Standard"]
print(standard_table)from Bio.Data import CodonTable
standard_table = CodonTable.unambiguous_dna_by_id[1]
print(standard_table)둘 다 같은 테이블을 호출하는건데, 위에껀 이름으로 부르고 밑에껀 ID로 부른다. 이름이나 ID는 위에 올린 NCBI 주소로 가면 나오니까 그걸로 부르면 된다.
Table 1 Standard, SGC0
| T | C | A | G |
--+---------+---------+---------+---------+--
T | TTT F | TCT S | TAT Y | TGT C | T
T | TTC F | TCC S | TAC Y | TGC C | C
T | TTA L | TCA S | TAA Stop| TGA Stop| A
T | TTG L(s)| TCG S | TAG Stop| TGG W | G
--+---------+---------+---------+---------+--
C | CTT L | CCT P | CAT H | CGT R | T
C | CTC L | CCC P | CAC H | CGC R | C
C | CTA L | CCA P | CAA Q | CGA R | A
C | CTG L(s)| CCG P | CAG Q | CGG R | G
--+---------+---------+---------+---------+--
A | ATT I | ACT T | AAT N | AGT S | T
A | ATC I | ACC T | AAC N | AGC S | C
A | ATA I | ACA T | AAA K | AGA R | A
A | ATG M(s)| ACG T | AAG K | AGG R | G
--+---------+---------+---------+---------+--
G | GTT V | GCT A | GAT D | GGT G | T
G | GTC V | GCC A | GAC D | GGC G | C
G | GTA V | GCA A | GAA E | GGA G | A
G | GTG V | GCG A | GAG E | GGG G | G
--+---------+---------+---------+---------+--아무튼 소환했다.
색인
저거 검색도 된다. 실화다.
print(standard_table.stop_codons)
['TAA', 'TAG', 'TGA']해당 테이블의 Stop codon을 검색하거나
print(mito_table.start_codons)
['ATT', 'ATC', 'ATA', 'ATG', 'GTG']개시코돈을 검색하거나(저거 2번 테이블이다)
print(standard_table.forward_table['AAA'])
K특정 코돈이 지정하는 아미노산을 찾을 수 있다.
'Coding > Python' 카테고리의 다른 글
| 번외편-코딩테스트 풀이 (0) | 2022.08.20 |
|---|---|
| Biopython으로 MSA 해보기 (0) | 2022.08.20 |
| Biopython SeqIO 써보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 레코드 생성하고 만져보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 가져오기 (0) | 2022.08.20 |
참고로 다른 코딩글은 여기다가 안 올린다. 코드블럭도 없고 카테고리도 없어서 어지간한 코딩 이야기는 노션과 워드프레스에 올리는 중... 아니면 미디움이나. 근데 이건 우째도 올렸네? 아 생물학 카테고리가 있잖아요.
Biopython은 생물정보학에서 쓰는건데 마침 심심하던 차에 잘됐다 써봐야징 해서 어제 깔았다. 리눅스의 경우 pip install bio(안되면 biopython)으로 걍 터미널에서 깔면 된다(우분투 20.02 LTS 기준). 윈도우마냥 pip 있는 데 안 찾아가도 됨 ㄹㅇ 편함. 근데 파이참 바로가기 없는 건 너무한 거 아니냐고... 내가 터미널 열고 직접 모시러 가야것냐...
*Notes: Biopython을 사용하려면 사용할 기능에 맞는 모듈을 모셔와야 한다. 코드에 그게 생략되는 경우가 있는데(부분삽입의 경우) from ~ import ~가 생략된 것이다.
일단 오늘의 시범조교를 모셔보자.
ATGGTAGCTCAAGCTCCTCATGATGATCATCAGGATGATGAGAAATTAGCAGCAGCGAGACAAAAAGAGATCGAAGATTGGTTACCAATTACTTCATCAAGAAATGCAAAGTGGTGGTACTCTGCTTTTCACAATGTCACCGCCATGGTCGGTGCCGGAGTTCTTGGACTCCCTTACGCCATGTCTCAGCTCGGATGGGGACCGGGAATTGCAGTGTTGGTTTTGTCATGGGTCATAACACTATACACATTATGGCAAATGGTGGAAATGCATGAAATGGTTCCTGGAAAGCGTTTTGATCGTTACCATGAGCTCGGACAACACGCGTTTGGAGAAAAACTCGGTCTTTATATCGTTGTGCCGCAACAATTGATCGTTGAAATCGGTGTTTGCATCGTTTATATGGTCACTGGAGGCAAATCTTTAAAGAAATTTCATGAGCTTGTTTGTGATGATTGTAAACCAATCAAGCTTACTTATTTCATCATGATCTTTGCTTCGGTTCACTTCGTCCTCTCTCATCTTCCTAATTTCAATTCCATCTCCGGCGTTTCTCTTGCTGCTGCCGTTATGTCTCTCAGCTACTCAACAATCGCATGGGCATCATCAGCAAGCAAAGGTGTTCAAGAAGACGTTCAATACGGTTACAAAGCGAAAACAACAGCCGGTACGGTTTTCAATTTCTTCAGCGGTTTAGGTGATGTGGCATTTGCTTACGCGGGTCATAATGTGGTCCTTGAGATCCAAGCAACTATCCCTTCAACGCCTGAGAAACCCTCAAAAGGTCCCATGTGGAGAGGAGTCATCGTTGCTTACATCGTCGTAGCGCTCTGTTATTTCCCCGTGGCTCTCGTTGGATACTACATTTTCGGGAACGGAGTCGAAGATAATATTCTCATGTCACTTAAGAAACCGGCGTGGTTAATCGCCACGGCGAACATCTTCGTCGTGATCCATGTCATTGGTAGTTACCAGATATATGCAATGCCGGTATTTGATATGATGGAGACTTTATTGGTCAAGAAGCTAAATTTCAGACCAACCACAACTCTTCGGTTCTTTGTCCGTAATTTCTACGTTGCTGCAACTATGTTTGTTGGTATGACGTTTCCGTTCTTCGGTGGGCTTTTGGCGTTCTTTGGTGGATTCGCGTTTGCCCCAACCACATACTTCCTCCCTTGCGTCATTTGGTTAGCCATCTACAAACCCAAGAAATACAGCTTGTCTTGGTGGGCCAACTGGGTATGCATCGTGTTTGGTCTTTTCTTGATGGTCCTATCGCCAATTGGAGGGCTAAGGACAATCGTTATTCAAGCAAAAGGATACAAGTTTTACTCATAA
At5g40780의 CDS 시퀀스이다. (코드블럭은 있는데 wrap이 안돼서 걍 생으로 붙였음)
이걸 시퀀스 오브젝트로 생성하는 방법도 간단하다. 그냥 모듈 모셔온 다음 Seq=("")로 감싸버리면 된다.
at5g40780=Seq("ATGGTAGCTCAAGCTCCTCATGATGATCATCAGGATGATGAGAAATTAGCAGCAGCGAGACAAAAAGAGATCGAAGATTGGTTACCAATTACTTCATCAAGAAATGCAAAGTGGTGGTACTCTGCTTTTCACAATGTCACCGCCATGGTCGGTGCCGGAGTTCTTGGACTCCCTTACGCCATGTCTCAGCTCGGATGGGGACCGGGAATTGCAGTGTTGGTTTTGTCATGGGTCATAACACTATACACATTATGGCAAATGGTGGAAATGCATGAAATGGTTCCTGGAAAGCGTTTTGATCGTTACCATGAGCTCGGACAACACGCGTTTGGAGAAAAACTCGGTCTTTATATCGTTGTGCCGCAACAATTGATCGTTGAAATCGGTGTTTGCATCGTTTATATGGTCACTGGAGGCAAATCTTTAAAGAAATTTCATGAGCTTGTTTGTGATGATTGTAAACCAATCAAGCTTACTTATTTCATCATGATCTTTGCTTCGGTTCACTTCGTCCTCTCTCATCTTCCTAATTTCAATTCCATCTCCGGCGTTTCTCTTGCTGCTGCCGTTATGTCTCTCAGCTACTCAACAATCGCATGGGCATCATCAGCAAGCAAAGGTGTTCAAGAAGACGTTCAATACGGTTACAAAGCGAAAACAACAGCCGGTACGGTTTTCAATTTCTTCAGCGGTTTAGGTGATGTGGCATTTGCTTACGCGGGTCATAATGTGGTCCTTGAGATCCAAGCAACTATCCCTTCAACGCCTGAGAAACCCTCAAAAGGTCCCATGTGGAGAGGAGTCATCGTTGCTTACATCGTCGTAGCGCTCTGTTATTTCCCCGTGGCTCTCGTTGGATACTACATTTTCGGGAACGGAGTCGAAGATAATATTCTCATGTCACTTAAGAAACCGGCGTGGTTAATCGCCACGGCGAACATCTTCGTCGTGATCCATGTCATTGGTAGTTACCAGATATATGCAATGCCGGTATTTGATATGATGGAGACTTTATTGGTCAAGAAGCTAAATTTCAGACCAACCACAACTCTTCGGTTCTTTGTCCGTAATTTCTACGTTGCTGCAACTATGTTTGTTGGTATGACGTTTCCGTTCTTCGGTGGGCTTTTGGCGTTCTTTGGTGGATTCGCGTTTGCCCCAACCACATACTTCCTCCCTTGCGTCATTTGGTTAGCCATCTACAAACCCAAGAAATACAGCTTGTCTTGGTGGGCCAACTGGGTATGCATCGTGTTTGGTCTTTTCTTGATGGTCCTATCGCCAATTGGAGGGCTAAGGACAATCGTTATTCAAGCAAAAGGATACAAGTTTTACTCATAA")
print(at5g40780)이런 식으로.
print(type(at5g40780))
<class 'Bio.Seq.Seq'>누가봐도 시퀀스다.
엥 근데 저거 걍 텍스트랑 뭔 차이임? Biopython을 통해 시퀀스로 생성한 데이터는 complement sequence와 reverse complement sequence를 만들 수 있다. 즉, 시퀀스로 불러온 CDS에 상보적인 시퀀스를 명령어 한 방에 만들 수 있는 것이다. 텍스트는 저런거 안된다.
from Bio.Seq import Seq
at5g40780=Seq("ATGGTAGCTCAAGCTCCTCATGATGATCATCAGGATGATGAGAAATTAGCAGCAGCGAGACAAAAAGAGATCGAAGATTGGTTACCAATTACTTCATCAAGAAATGCAAAGTGGT")
print("Sequence",at5g40780)
print("Complement sequence",at5g40780.complement())
print("Reverse complement sequence",at5g40780.reverse_complement())이렇게 입력하면
Sequence ATGGTAGCTCAAGCTCCTCATGATGATCATCAGGATGATGAGAAATTAGCAGCAGCGAGACAAAAAGAGATCGAAGATTGGTTACCAATTACTTCATCAAGAAATGCAAAGTGGT
Complement sequence TACCATCGAGTTCGAGGAGTACTACTAGTAGTCCTACTACTCTTTAATCGTCGTCGCTCTGTTTTTCTCTAGCTTCTAACCAATGGTTAATGAAGTAGTTCTTTACGTTTCACCA
Reverse complement sequence ACCACTTTGCATTTCTTGATGAAGTAATTGGTAACCAATCTTCGATCTCTTTTTGTCTCGCTGCTGCTAATTTCTCATCATCCTGATGATCATCATGAGGAGCTTGAGCTACCAT짜자잔
complement는 CDS 시퀀스에 그냥 상보적인거고, reverse complement는 그 상보적인 시퀀스 순서를 뒤집은 모양이다.
아니 근데 DNA가 겁나 바이트가 길어요... 저 시범조교 CDS도 1300자가 넘는데 어느 세월에 그거 복붙할거임... 그래서 FASTA와 Genbank의 데이터를 직접 열어볼 것이다.
FASTA 시범조교는 PDB ID 2B10, cytochrome c peroxiase이다.
>2B10_1|Chains A, C|Cytochrome c peroxidase, mitochondrial|Saccharomyces cerevisiae (4932)
TTPLVHVASVEKGRSYEDFQKVYNAIALKLREDDEYDNYIGYGPVLVRLAWHTSGTWDKHDNTGGSYGGTYRFKKEFNDPSNAGLQNGFKFLEPIHKEFPWISSGDLFSLGGVTAVQEMQGPKIPWRCGRVDTPEDTTPDNGRLPDADKDADYVRTFFQRLNMNDREVVALMGAHALGKTHLKNSGYEGPWGAANNVFTNEFYLNLLNEDWKLEKNDANNEQWDSKSGYMMLPTDYSLIQDPKYLSIVKEYANDQDKFFKDFSKAFEKLLENGITFPKDAPSPFIFKTLEEQGL
>2B10_2|Chains B, D|Cytochrome c iso-1|Saccharomyces cerevisiae (4932)
TEFKAGSAKKGATLFKTRCLQCHTVEKGGPHKVGPNLHGIFGRHSGQAEGYSYTDANIKKNVLWDENNMSEYLTNPKKYIPGTKMASGGLKKEKDRNDLITYLKKACEFASTA 파일 형식은 이렇게 생겼다. 일단 저 파일을 어떻게 가져올거냐... PDB 가서 2B10으로 검색하고 FASTA 파일을 받아보자.
for seq_record in SeqIO.parse("/home/koreanraichu/rcsb_pdb_2B10.fasta","fasta"):
print(seq_record.id)
print(repr(seq_record.seq))
print(len(seq_record))
2B10_1|Chains
Seq('TTPLVHVASVEKGRSYEDFQKVYNAIALKLREDDEYDNYIGYGPVLVRLAWHTS...QGL')
294
2B10_2|Chains
Seq('TEFKAGSAKKGATLFKTRCLQCHTVEKGGPHKVGPNLHGIFGRHSGQAEGYSYT...ACE')
108FASTA 파일은 이런 식으로 가져올 수 있다. (seqIO 데려오면 된다) 예? 경로요? 아니 이거 리눅슨디?
사실 분량때문에 넣지는 못하지만
print(seq_record)만 치면 전체 파일을 볼 수 있다. 처음 하시는 분들은 전체를 보고 저 예제를 따라해보는 걸 추천한다.
for seq_record in SeqIO.parse("/home/koreanraichu/sequence.gb","genbank"):
print(seq_record.id)
print(repr(seq_record.seq))
print(len(seq_record))Genbank에서 뭐 검색하고 그거 gk(gbk)파일로 받으면 그게 genbank 파일이다. 이것도 일단 다운받아서 불러와야 한다. 웹에 있는거요? 그거 바로 못갖고온다.
아무튼 저 코드를 실행하면
NM_000517.6
Seq('ACTCTTCTGGTCCCCACAGACTCAGAGAGAACCCACCATGGTGCTGTCTCCTGC...GCA')
576이렇게 된다. 그리고 얘도 FASTA와 마찬가지로
print(seq_record)를 주면 전체 파일을 볼 수는 있는데 분량이 어유...
'Coding > Python' 카테고리의 다른 글
| 번외편-코딩테스트 풀이 (0) | 2022.08.20 |
|---|---|
| Biopython으로 MSA 해보기 (0) | 2022.08.20 |
| Biopython SeqIO 써보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 레코드 생성하고 만져보기 (0) | 2022.08.20 |
| Biopython으로 시퀀스 다뤄보기 (0) | 2022.08.20 |